
What’s Causing a Woman’s Intertriginous Abscesses With Malodorous Discharge?
Authors:
Alexander K. C. Leung, MD, and Benjamin Barankin, MD
Citation:
Leung AKC, Barankin B. What’s causing a woman’s intertriginous abscesses with malodorous discharge? [Published online November 16, 2016]. Consultant360.

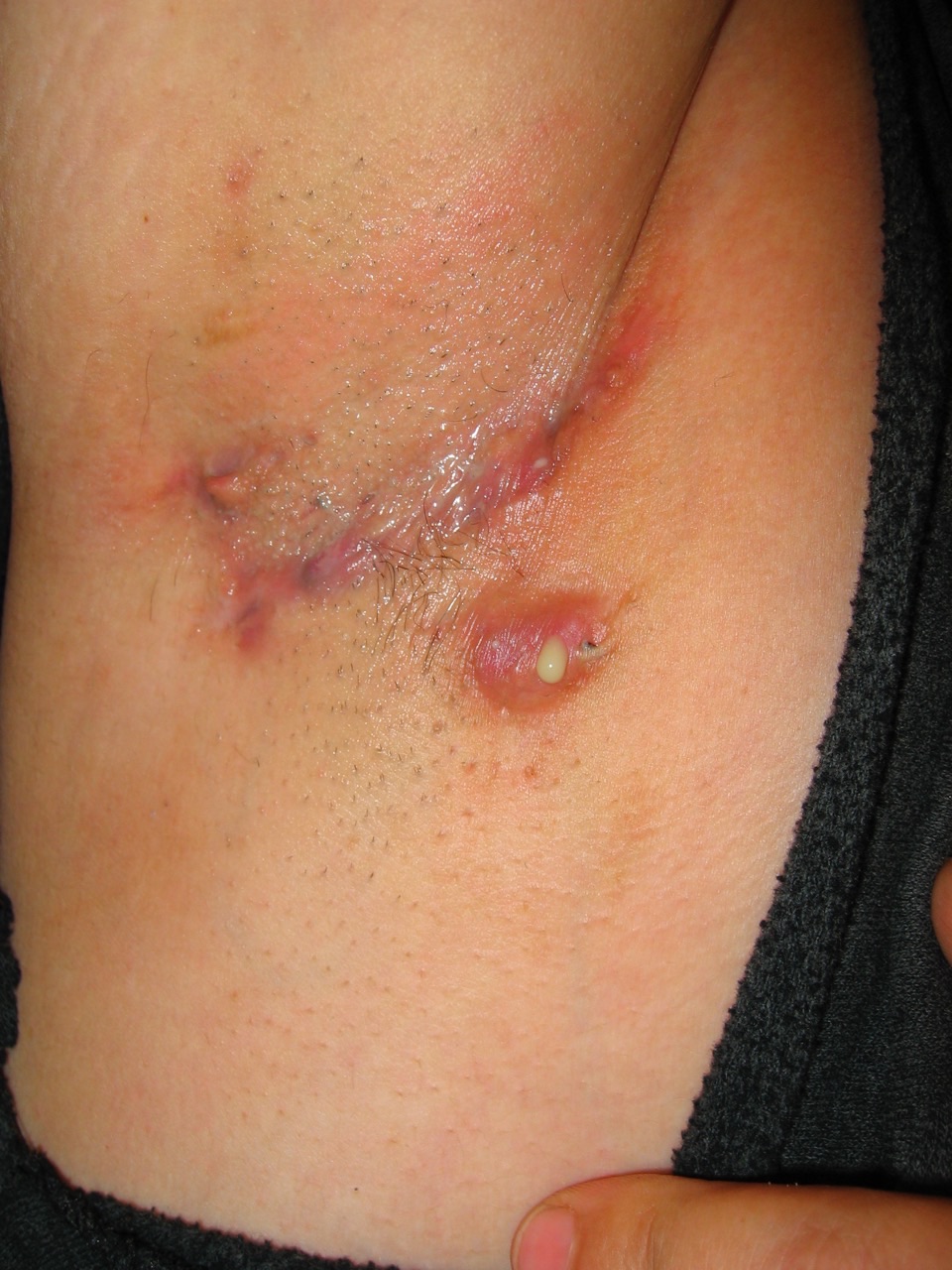
A 28-year-old white woman presented with recurrent boils in the axillary and inguinal areas. The lesions had been fluctuating in severity for the preceding 5 or 6 years and often had flared premenstrually. She had been treated with short courses of oral antibiotics from time to time with modest success. She smoked 1 pack of cigarettes a day. She had mild hypertension and borderline hypercholesterolemia. There was no family history of similar skin disorders.
On physical examination, the patient was noted to be obese, with a body mass index of 41 mg/kg2. Her pulse was 76 beats/min, and her blood pressure was 140/85 mm Hg. There were multiple scars in both axillae and inguinal regions, and an indurated abscess in the right axillary area with a foul-smelling, purulent discharge.

What's Your Diagnosis?
- Folliculitis
- Infected epidermoid cysts
- Erysipelas
- Hidradenitis suppurativa
Answer on next page.
Answer: Hidradenitis suppurativa
Hidradenitis suppurativa (HS), also known as acne inversa or apocrine acne, is a chronic, recurrent, inflammatory disorder of pilosebaceous units that is characterized by recurrent, painful, subcutaneous nodules and/or abscesses affecting primarily the apocrine gland-bearing areas of the body, notably the intertriginous areas.1 The condition was first described in 1854 by Aristide Verneuil, a French surgeon. Hence, the disease also is referred to as Verneuil disease.2
EPIDEMIOLOGY
The overall prevalence is estimated at 0.05% to 1%.3-5 The condition most commonly develops in individuals in the second or third decades of life, with peak severity occurring 10 to 15 years after onset of symptoms.1,4 In female patients, the onset occurs almost always after menarche and before menopause.1,6,7 Premenstrual flares are commonly reported.8 The female to male ratio is 3 to 1.6,9 The condition is most common in patients of African descent.4,10,11
ETIOPATHOGENESIS
HS is believed to result from follicular occlusion of the pilosebaceous unit by infundibular hyperkeratosis, leading to ductal dilatation and stasis in the glandular component.10,11 Secondary rupture of the infundibular duct results in leakage of contents into the surrounding tissue, leading to local inflammation and stimulation of the immune system.5,12 Altered innate and adaptive immune systems with upregulation of potent inflammatory cytokines (such as interleukins 1β and 10 and tumor necrosis factor α) accelerate the destruction of the portion of the pilosebaceous unit below the site of rupture.
Hormonal overstimulation may play a role in ductal keratinocyte proliferation. Given that the serum androgens are not elevated in the majority of patients with HS, end-organ sensitivity likely is responsible.12 Bacterial infection does not have a major role in the pathogenesis, as evidenced by the sterility of pus and negative bacterial cultures from early lesions. More likely, bacteria isolated in late lesions are colonizers or secondary invaders.12
There is a genetic predisposition, as one-third of patients have a family history of the disease.13 An autosomal dominant mode of inheritance has been reported.12,13 In a small number of cases, the disease has been linked to chromosomal bands 1p21.1 through 1q25.3 and mutations of the γ-secretase complex.8,12 Predisposing factors include smoking, obesity, mechanical stress (pressure, friction) on skin, hot and humid environment, and metabolic syndrome (obesity, hypertension, diabetes, and dyslipidemia).14-16 Conditions associated with HS include severe acne (acne conglobata), psoriasis, pyoderma gangrenosum, ulcerative colitis, and Crohn disease.4,12,16
HISTOPATHOLOGY
Histologic findings vary according to the stage of the disease and include infundibular spongiosis, follicular hyperkeratosis, hyperplasia of the follicular epithelium, lymphocytic perifolliculitis, comedo formation, follicular rupture, granulation tissue, foreign-body giant cells, dermal/subcutaneous abscess, fibrosis, and sinus tract lined by stratified squamous epithelium.9
NEXT: Clinical Manifestations
CLINICAL MANIFESTATIONS
The mean age of onset is approximately 21 years.14 Early onset may be associated with a greater risk of widespread disease.12 Typically, HS presents with painful, deep-seated, inflamed nodules. The nodules are situated in the deeper dermis and therefore appear rounded instead of having a pointed, purulent appearance or the central necrosis of simple boils.8,13 Approximately 50% of patients experience prodromal symptoms of warmth, burning, stinging, hyperhidrosis, pain, and/or pruritus 1 to 2 days before overt nodules develop.8 The nodules may spontaneously resolve, rupture, or coalesce to form deep, tender dermal abscesses. Rupture of a nodule/abscess may result in malodorous purulent discharge. There is generally no associated fever or regional lymphadenopathy.8,9,17
With time, the affected skin may be left with plaque-like induration, hypertrophic or ropelike scars, and sometimes dermal contractures.1 Double-ended or bridged scars and “tombstone” open comedones in burnout regions are characteristic.5 Chronic or recurrent lesions may result in sinus tract and/or fistula formation.
Sites of predilection include axillary, inguinal, perianal, and perianal areas, and, in women, inframammary areas, with the axilla being the most common site.12 There is a tendency for lesions to recur at the same sites or at nearby sites.14 Over time, more sites are affected.
The Hurley classification system, which has been widely used to stratify disease severity, is as follows18:
- Hurley stage I: Abscess formation (single or multiple) without sinus tracts and cicatrization.
- Hurley stage II: Recurrent abscesses with tract formation and cicatrization, single or multiple widely separated lesions.
- Hurley stage III: Diffuse or near-diffuse involvement or multiple interconnected tracts and abscesses across the entire area.
DIAGNOSIS AND DIFFERENTIAL DIAGNOSIS
The diagnosis is usually clinical, based on typical lesions (painful deep-seated inflammatory nodules, comedones, abscesses, sinus tracts, fibrotic scars), typical locations (intertriginous areas, often bilateral), and typical course (relapses and chronicity). A positive family history and a history of premenstrual flares may aid in the diagnosis.8 A skin biopsy is necessary only if the diagnosis is in doubt.
Routine bacterial cultures generally are negative and are not indicated. They should be performed if secondary infection is suspected. Ultrasonography is helpful in identifying subclinical extension of the lesions if extensive surgery is planned.13
The differential diagnosis includes carbuncles, furuncles, tuberculosis, infected epidermoid cysts, erysipelas, erythrasma, infected Bartholin glands, infected pilonidal cysts, lymphogranuloma venereum, scrofuloderma, granuloma inguinale, and Crohn disease.
COMPLICATIONS
HS may result in repeated absences from work or school because of pain, malodorous discharge, or unsightly appearance.7 The disease has an adverse effect on the quality of life because of its chronic and recurrent nature, the lack of satisfying treatment options, and social isolation. Affected patients may experience feelings of sadness, shame, low self-esteem, sexual disharmony, and depression.7,17 Contractures may result from ropelike scars. Fistula into the bladder, urethra, rectum, and anus, as well as reactive arthritis and interstitial keratitis, are potential complications.12 Occasionally, extensive scarring may result in lymphatic obstruction, lymphangiectasia, and lymphedema.9 Rarely, patients with longstanding disease may develop squamous cell carcinoma, buccal carcinoma, or hepatocellular carcinoma.9,13
PROGNOSIS AND MANAGEMENT
HS is a debilitating disease that tends to run a chronic course with relapses. The mean duration of the active disease is approximately 19 years.8,19
Smoking cessation, weight control, and avoidance of tight-fitting clothing are beneficial aspects of general management. Analgesics may have to be given for pain control. Psychological support should be offered if necessary.
Treatment should be individualized, taking into consideration of the severity and distribution of the lesions, the subjective impact on quality of life, and the risks and benefits of any treatment. Treatment options for Hurley stage I disease include topical antibiotics (eg, clindamycin, fusidic acid), a short course of systemic antibiotics (eg, tetracycline, dapsone, clindamycin-rifampicin), intralesional corticosteroids, and surgical procedures.6,7,12,17 Intralesional corticosteroids should be considered for rapid reduction with acute flares and resolution of early, painful, inflammatory lesions, as well as for the treatment of recalcitrant nodules and sinus tracts.11,12 They can be used as monotherapy or as an adjunct to systemic therapies. Punch debridement (mini deroofing) can be used to evacuate a new inflamed nodule.12 Abscesses that are fluctuant and painful may need incision and drainage.
Treatment options for Hurley stage II disease include long-term (3 months or more) systemic antibiotics (eg, doxycycline, dapsone, tetracycline, amoxicillin-clavulanic acid, clindamycin-rifampicin, rifampicin-moxifloxacin-metronidazole), intralesional corticosteroids, laser ablation (eg, carbon dioxide laser, neodymium-doped yttrium aluminum garnet laser), limited surgical excision (eg, deroofing of an abscess or nodule, exteriorization of sinus tracts) and, in women, antiandrogenic agents (eg, ethinyl estradiol-norgestrel, finasteride, cyproterone acetate).7,12,17
Wide and radical excision of the lesions and surrounding apocrine gland-bearing areas (delineated by the hairy surfaces of the affected regions) down to soft, normal tissue with healing by secondary intention or grafting is the gold standard for Hurley stage III disease.7,10,17 Tumor necrosis factor α inhibitors (eg, infliximab, adalimumab) may be considered as an adjunct therapy to improve the quality of life.6,17
Alexander K. C. Leung, MD, is clinical professor of pediatrics at the University of Calgary and a pediatric consultant at the Alberta Children’s Hospital in Calgary, Alberta, Canada.
Benjamin Barankin, MD, is a dermatologist and the medical director and founder of the Toronto Dermatology Centre in Toronto, Ontario, Canada.
REFERENCES:
- Li K, Barankin N. Can you identify this condition? Hidradenitis suppurativa. Can Fam Physician. 2011;57(9):1023-1024.
- Verneuil A. Études sur les tumeurs de la peau; de quelques maladies des glandes sudoripares. Arch Gen Med. 1854;4:447-468, 693-705.
- Cosmatos I, Matcho A, Weinstein R, Montgomery MO, Stang P. Analysis of patient claims data to determine the prevalence of hidradenitis suppurativa in the United States. J Am Acad Dermatol. 2013;68(3):412-419.
- Shlyankevich J, Chen AJ, Kim GE, Kimball AB. Hidradenitis suppurativa is a systemic disease with substantial comorbidity burden: a chart-verified case-control analysis. J Am Acad Dermatol. 2014;71(6):1144-1150.
- Yazdanyar S, Jemec GBE. Hidradenitis suppurativa: a review of cause and treatment. Curr Opin Infect Dis. 2011;24(2):118-123.
- Hughes R, Kelly G, Sweeny C, Lally A, Kirby B. The medical and laser management of hidradenitis suppurativa. Am J Clin Dermatol. 2015;16(2):111-123.
- Schulman ML, Pantuso HM. Hidradenitis suppurativa: a case study. J Pediatr Health Care. 2015;29(4):364-370.
- Revuz J. Hidradenitis suppurativa. J Eur Acad Dermatol Venereol. 2009;23(9):985-998.
- Lee RA. Hidradenitis suppurativa (acne inversa). Clinical Decision Support in Medicine: Dermatology. http://www.decisionsupportinmedicine.com. Accessed July 6, 2016.
- Buimer MG, Wobbes T, Klinkenbijl JHG. Hidradenitis suppurativa. Br J Surg. 2009;96(4):350-360.
- Wollina U, Koch A, Heing B, Kittner T, Nowak A. Acne inversa (hidradenitis suppurativa): a review with a focus on pathogenesis and treatment. Indian Dermatol Online J. 2013;4(1):2-11.
- Margesson LJ, Danby FW. Hidradenitis suppurativa (acne inversa): pathogenesis, clinical features, and diagnosis. UpToDate. http://www.uptodate.com/contents/hidradenitis-suppurativa-acne-inversa-pathogenesis-clinical-features-and-diagnosis. Updated March 10, 2016. Accessed July 6, 2016.
- Jemec GBE. Hidradenitis suppurativa. N Engl J Med. 2012;366(2):158-164.
- Collier F, Smith RC, Morton CA. Diagnosis and management of hidradenitis suppurativa. BMJ. 2013;346:f2121.
- Gold DA, Reeder VJ, Mahan MG, Hamzavi IH. The prevalence of metabolic syndrome in patients with hidradenitis suppurativa. J Am Acad Dermatol. 2014;70(4):699-703.
- Pascoe VL, Kimball AB. Hidradenitis suppurativa: current progress and future questions. JAMA Dermatol. 2014;150(12):1263-1264.
- Zouboulis CC, Desai N, Emtestam L, et al. European S1 guideline for the treatment of hidradenitis suppurativa/acne inversa. J Eur Acad Dermatol Venereol. 2015;29(4):619-644.
- Hurley HJ. Axillary hyperhidrosis, apocrine bromhidrosis, hidradenitis suppurativa, and familial benign pemphigus: surgical approach. In: Roenigk RK, Roenigk HH Jr, eds. Roenigk and Roenigk’s Dermatologic Surgery: Principles and Practice. 2nd ed. New York, NY: Marcel Dekker; 1996:623-645.
- von der Werth JM, Williams HC. The natural history of hidradenitis suppurativa. J Eur Acad Dermatol Venereol. 2000;14(5):389-392.