
A Young Woman with Sharp, Constant Pain in Her Abdomen
A 36-year-old female was admitted to the hospital with a 3-day history of nausea, emesis, and abdominal pain. She noted sharp, constant pain in her epigastric region with no identifiable triggers. The patient denied fevers, chills, diarrhea, or dysuria.
History
Her past medical history is significant for 15 pregnancies (the patient is currently 1-month postpartum) and an ectopic pregnancy treated with right salpingectomy. She was on no home medications at the time of admission.
She reported smoking half-a-pack daily of past 10 to 15 years and also admitted to social alcohol consumption; she had several drinks 4 days prior. She denies any drug use.
Review of systems was negative for dyspnea, chest pain, and paroxysmal nocturnal dyspnea but positive for new onset lower extremity edema.
Physical ExamINATION
On admission, the patient was afebrile, not tachycardic or tachypneic. She had epigastric abdominal tenderness and mild 1-2+ pitting edema of bilateral extremities to the lower shins. The patient also demonstrated mild scleral icterus and bilateral basilar crackles on pulmonary examination. The neurologic exam was nonfocal. The cardiac exam revealed regular rate and rhythm without any murmurs, gallops, or rubs appreciated.
Laboratory Tests
Admission labs showed serum creatinine levels at 0.9 mg/dL, blood urea nitrogen at 7 mg/dL, lipase at 112 U/L, alkaline phosphatase at 161 U/L, aspartate aminotransferase of 37 U/L, alanine transaminase of 17 U/L, and total bilirubin of 1.1 mg/dL (direct bilirubin of 0.5 mg/dL). Her complete blood count showed white blood cells at 5.4 K/mm3, platelets at 349 K/mm3, and hemoglobin at 11.7 g/dL.
A CT scan of the abdomen and pelvis revealed edema of pancreatic head and body, with surrounding fat stranding.
Initial Treatment
The patient was admitted with a diagnosis of pancreatitis and started on intravenous fluids and a pain regimen to support her through her pancreatitis. However, during the next 3 days, her creatinine levels progressively worsened to 1.7 mg/dL, her hemoglobin dropped to 5.9 g/dL, and she had a drop in her platelet count to 106 K/mm3.
Hematology was consulted and upon reviewing her peripheral smear, she was found to have ≥6 schistocytes per high-powered field exam (Figure 1), thrombocytopenia with platelets down to 50 K/mm3, worsening renal function with a creatinine level close to 3 mg/dL, and worsening generalized edema.
ADAMTS 13 level was checked and came back as <10%.
What is the most appropriate next step in the management of this patient?
A. Arrange for infusion of intravenous immunoglobulin.
B. Arrange for immediate plasma exchange.
C. Start therapy with eculizumab.
D. Continue supportive measures and closely monitor patient’s counts.
(Answer and discussion on next page)
Correct Answer: B
The patient’s clinical history, symptoms, and peripheral smear strongly suggest a diagnosis of thrombotic thrombocytopenic purpura (TTP)—a rare but deadly disorder, characterized classically by microangiopathic hemolytic anemia, thrombocytopenia, variable degrees of renal insufficiency, neurologic impairment, and other organ failure.1 It is among a group of disorders—which also include atypical complement-mediated thrombotic microangiopathy and hemolytic uremic syndrome (HUS)—that are referred to as thrombotic microagiopathies and are characterized by microangiopathic hemolytic anemia, thrombocytopenia, and organ injury.2
The pathophysiologic hallmark of TTP is the presence of large von Willebrand factor (vWF) multimers and low levels of ADAMTS13, the protease that normally cleaves vWF multimers into the “right size.”2 In the absence of adequate ADAMTS13 activity, abnormally large vWF multimers are more easily accumulated and cause intravascular vWF-platelet aggregation and microvascular thrombosis.3
In the event that a diagnosis of TTP is strongly suspected, even if all 5 traditional features are not present, arrangements should be made to start plasma exchange on the patient. Thus, Answer B is correct. Prior to the utilization of plasma exchange, TTP was associated with dismal outcomes with an overall survival rate of only 10%. However, a 1991 randomized trial found that the incorporation of plasma exchange into the treatment algorithm for TTP can increase the overall survival rate to approximately 79%.2
In theory, the aim of plasma exchange is to replenish deficient stores of ADAMTS13 and remove autoantibodies directed against ADAMTS13.4 However, even in patients with normal or only slightly depressed ADAMTS13 levels (traditionally TTP is characterized by an ADAMTS13 level <10%), plasma exchange appears to have a significant impact in improving overall clinical outcomes by providing normal vWF, which competes with and diminishes the effect of the enlarged vWF multimers, and removing the large vWF multimers themselves.5
Differential Diagnosis
Given the significant impact of early intervention with plasma exchange, Answer D would not be an appropriate course of action for this patient.
The low platelet count in our patient may raise the possibility of a diagnosis of immune thrombocytopenic purpura (ITP). However, the patient’s clinical presentation and peripheral smear would argue against such a diagnosis. In ITP, the patient’s peripheral smear is characterized by the absence of platelets in the presence of normal counts of red and white blood cells with near normal to normal morphology. Furthermore, schistocytes, especially at as high a number per high power field as seen with our patient (Figure 1), and an association with acute onset renal failure would not be encountered in patients with ITP. Therefore, Answer A—intravenous immunoglobulin that is often utilized in ITP—would not be an appropriate treatment choice for our patient.

Eculizumab is a recombinant humanized monoclonal antibody directed against C5 in the complement cascade, which has been previously approved for the management of paroxysmal nocturnal hemoglobinuria. More recently, based on the results of 2 pivotal single-arm prospective studies, eculizumab has recently received FDA approval for the treatment of complement-mediated HUS. Complement-mediated HUS is another microangiopathy syndrome that is characterized by normal ADAMTS13 levels, severe renal failure, and lack of response to plasma exchange.6 On reviewing our patient’s clinical picture, she has severely depressed ADAMTS13 levels and does not have a degree of renal failure usually encountered with complement-mediated HUS. Furthermore, confirmation of a diagnosis of complement-mediated HUS currently requires screening for mutations in the complement cascade and/or demonstration of failure of response with at least 5 days of plasma exchange.6 Therefore, Answer C would not be appropriate for our patient at this time.
Outcome of the Case
Arrangements were made with the blood bank to initiate plasma exchange and corticosteroids were started to address autoimmunity. There was prompt daily improvement in the platelet count, blood smear, lactate dehydrogenase (LDH), and hemoglobin. By day 6, platelets were 210,000/mm3, LDH was essentially normal, and serum creatinine was 0.9 mg/dL.
The patient was discharged and all parameters remained normal at her clinic visit 2 weeks later. A repeat ADAMTS13 level is pending to determine the pace of corticosteroid tapering and potential need for additional immune suppressive measures (eg, rituximab).
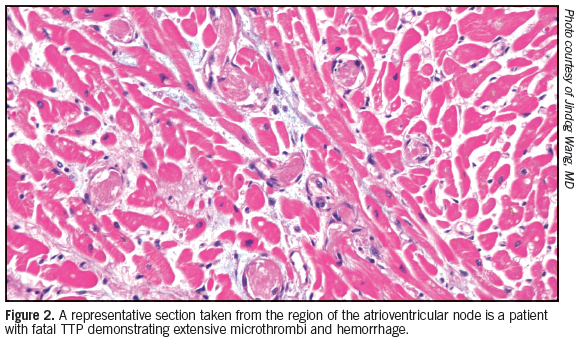
Take-Home Message
Thrombotic thrombocytopenic purpura is a microangiopathy syndrome classically characterized by the “pentad” of thrombocytopenia, microangiopathic hemolytic anemia, renal insufficiency, fever, and waxing/waning neurologic symptoms. It is now known to be an autoimmune disease caused by an autoantibody to the ADAMST13 protease that results in accumulation of abnormally large vWF in plasma capable of causing platelet microthrombi diffusely in the microcirculation. Prompt plasma exchange with adjuvant immune
suppression has resulted in a stunning improvement in prognosis from 85% mortality rate
to approximately the same degree of remission and long-term survivorship.
Nasheed M. Hossain, MD, is a fellow of hematology and oncology at Temple University Hospital and Fox Chase Cancer Center, both in Philadelphia, PA.
John Kaczmar, MD, is a fellow of hematology and oncology at Temple University Hospital and Fox Chase Cancer Center, both in Philadelphia, PA.
Ronald Rubin, MD, is a professor of medicine at Temple University School of Medicine and chief of clinical hematology in the department of medicine at Temple University Hospital, both in Philadelphia, PA.
References:
- Chaturvedi S, Carcioppolo D, Zhang L, McCrae K. Management and outcomes for patients with TTP: analysis of 100 cases at a single institution. Am J Hematol. 2013;88(7):560-565.
- George JN, Nester CM. Syndromes of thrombotic microangiopathy. N Engl J Med. 2014;371(7):654-666.
- Tsai HM. Untying the knot of thrombotic thrombocytopenic purpura and atypical hemolytic uremic syndrome. Am J Med. 2013;
- 126(3):200-209.
- George JN. How I treat patients with thrombotic thrombocytopenic purpura: 2010. Blood. 2010;116(20):4060-4069.
- Vesely SK, George JN, Lämmle B, et al. ADAMTS13 activity in thrombotic thrombocytopenic purpura-hemolytic uremic syndrome: relation to presenting features and clinical outcomes in a prospective cohort of 142 patients. Blood. 2003;102(1):60-68.
- Cataland SR, Wu HM. Atypical hemolytic uremic syndrome and thrombotic thrombocytopenic purpura: Clinically differentiating the thrombotic microagiopathies. Eur J Intern Med. 2013;24(6):486-491.