
Pulmonary Fibrosis: Diagnosis and Treatment
ABSTRACT: Interstitial lung diseases (ILDs) are a heterogeneous group of maladies with similar clinical presentation. Identification of a particular cause is challenging and the treating physicians should pay careful attention to the details in history and chest imaging. A multidisciplinary approach for diagnosis and treatment is favored. Idiopathic pulmonary fibrosis (IPF) is the most common ILD, with a chronic, progressive course requiring significant healthcare resources. New breakthrough treatments are now available that can potentially delay the progression of the disease. Patients with IPF may also have coexistent conditions that could cause significant additional morbidity and must be identified and treated. Patients at high risk of mortality should be referred for lung transplant.
Interstitial lung diseases (ILDs) are a complex group of more than 150 different disorders, characterized by inflammation and fibrosis of the pulmonary interstitium and parenchyma. Patients with chronic fibrosing ILDs may have similar clinical presentation, and a confident diagnosis requires careful attention to details in the history, clinical presentation, radiographic findings, and histopathology. Several drugs, connective tissue diseases, and granulomatous diseases can cause lung injury and scarring. Idiopathic interstitial pneumonias (IIPs) are a subset of ILD, where a specific etiology cannot be determined.
The American Thoracic Society (ATS) and the European Respiratory Society (ERS) recently updated the multidisciplinary classification of IIPs (Table 1). Major IIPs are now broken into chronic, smoking-related, and acute/subacute disease.1 Idiopathic pulmonary fibrosis (IPF) is the most common IIP, with an estimated incidence of 16.3 per 100,000 persons.2. Most ILD centers employ a multidisciplinary, clinico-radiographic-pathological discussion to improve the accuracy and diagnostic confidence, and to develop a treatment strategy for patients. Lately, new breakthrough treatment options have become available in treatment for IPF and a proper diagnosis is crucial for patients to be started on the appropriate treatment. This article summarizes the diagnostic approach, complications, and treatment options available for IPF through a case presentation.

Case history: A 66-year-old male presented with shortness of breath on minimal exertion and chronic cough for longer than 1 year. His symptoms had been progressing 6 months prior to his visit, but became worse over the last 2 weeks. His most recent work experience was within the airline industry, and he admitted to smoking a pack of cigarettes daily for the last 25 years. His oxygen saturation was 87% on room air, and further examination was significant for digital clubbing and bilateral rales. Chest x-ray showed increased interstitial markings bilaterally.
What is the next best step in management of this patient? Answer: High-resolution CT scan of chest
When evaluating patients with suspected ILD, a constellation of clinical findings, including the clinical context and evolution of the disease, and radiographic findings, may provide important clues to the diagnosis.3 Certain diseases such as connective tissue-related lung diseases (CT-ILD), sarcoidosis, and lymphangiolieomatosis (LAM) are more common in middle-aged patients, whereas, IPF is a disease of the elderly. Diseases like LAM are more prevalent among women, but pneumoconiosis and rheumatoid arthritis-related ILD is much more common in men. Smokers are more likely to have respiratory bronchiolitis-associated interstitial lung disease (RB-ILD), desquamative interstitial pneumonitis (DIP), pulmonary Langerhans cell histiocytosis (PLCH) or IPF. On the other hand, smokers usually do not develop hypersensitivity pneumonitis (HP) or sarcoidosis. Acute presentation (less than 3 weeks in duration) is usually related to infection, acute HP, acute eosinophilic pneumonias, or acute idiopathic interstitial pneumonia. In contrast, diseases such as sarcoidosis, chronic HP, CT-ILD, and IPF have a much more indolent course, often spanning months to years.
The differential diagnosis is broad and a high-resolution CT (HRCT) scan of the chest can provide detailed information and substantially narrow the differential diagnosis in suspected ILDs. The physician should focus on identifying the dominant pattern, distribution of pathology, and potential clues pointing toward a particular disease.
A predominant pattern of ground-glass opacities (GGO) on CT with basilar reticular markings is related with nonspecific interstitial pneumonia (NSIP). RB-ILD and DIP may have centrilobular nodules and GGO, whereas cryptogenic organizing pneumonia consists of patchy peripheral or peribronchial consolidations with GGO, and rare diseases such as idiopathic pleuro-parenchymal fibroelastosis may have upper lobe predominant pleural and subpleural reticular opacities.4

The 2011 evidence-based guidelines on IPF recognize a diagnosis of IPF based on HRCT findings of usual interstitial pneumonia (UIP) and absence of other criteria inconsistent with UIP (Table 2).5 In a study of 168 patients with suspected ILD, presence of honeycombing on HRCT indicated the presence of UIP with a sensitivity of 90% and specificity of 86%. UIP pattern was the most important factor predictive of mortality.6 The 5-year survival of patients diagnosed with having a UIP pattern on CT scan, or on histopathology after surgical lung biopsy, is comparable and dependent on honeycombing and fibrosis score on CT.7-9 Therefore, not all patients with suspected IPF need a surgical lung biopsy for diagnosis. A definitive UIP pattern on CT scan in the absence of other causes is suggestive of IPF. (Figure 1).
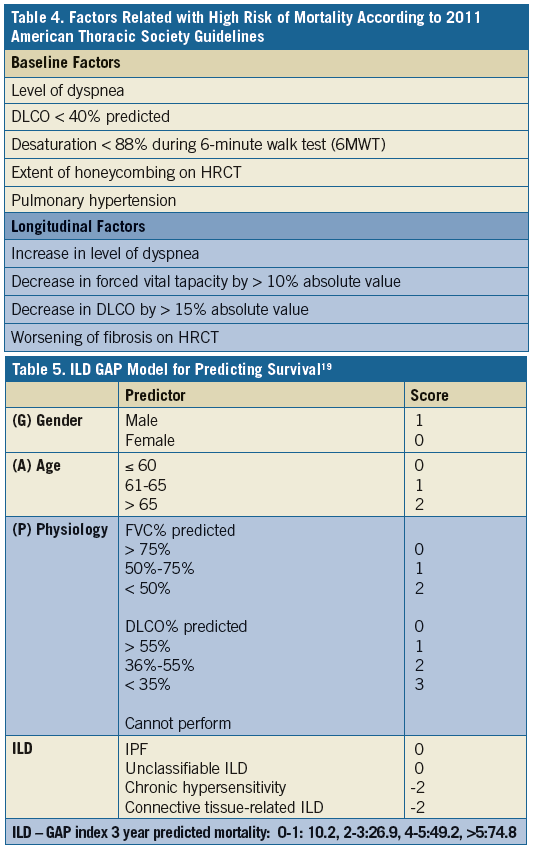
History continued: Patient had a CT scan of chest (Figure 2) and was interpreted as possible UIP pattern. A complete serology panel was negative for any associated connective tissue disease and he had no particular environmental exposure suggestive of HP. Patient underwent surgical lung biopsy.
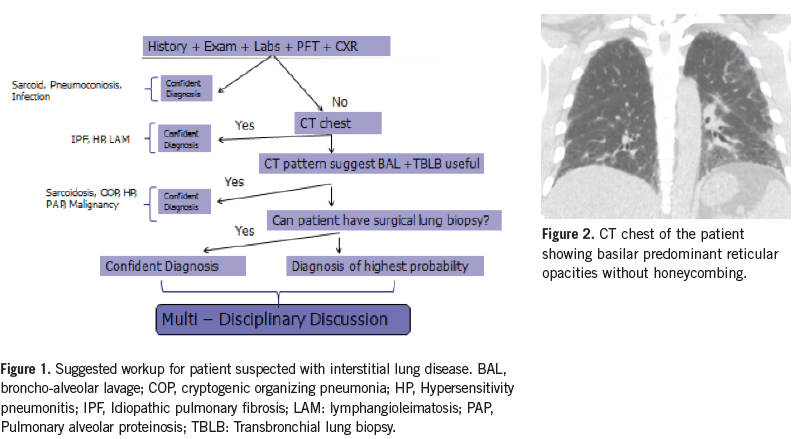
What is the next step in management of this patient? Answer: Multidisciplinary case discussion
Diagnosis of IPF requires exclusion of other possible etiologies and a UIP pattern on CT scan or histopathology (Table 3). There is limited role of transbronchial biopsy and brochoalveolar lavage in patients with UIP pattern; patients with possible UIP pattern should proceed with surgical lung biopsy if there is no contraindication and if the patient can tolerate it.5,10 Having a UIP pattern is associated with worse overall prognosis compared to NSIP pattern on lung biopsy as mentioned above.11 In patients undergoing lung biopsy, where different lobes were sampled and a concordant UIP pattern was identified, prognosis was worse. But even patients with discordant UIP pattern (UIP pattern on 1 lobe and NSIP pattern in the other lobe) had worse survival compared to concordant NSIP pattern.12,13 Because of obvious prognostic implications in patients with UIP pattern, those undergoing surgical lung biopsy should have multiple lobes sampled.

The diagnostic accuracy by clinicians alone and the level of agreement of individual diagnosis between radiologists and pathologists have been inconsistent on large multicenter trials on IPF patients.14 In a study with 58 suspected IIP patients, the diagnostic confidence and interobserver agreement improved, when the dynamic interaction between clinicians, radiologists, and pathologists increased.15 Current guidelines recommend that the clinician, radiologist, and pathologist interact in a multidisciplinary platform to establish a diagnosis of IIP.5
History continued: The patient was diagnosed with IPF. He has dyspnea with moderate exertion and was started on 2L oxygen via nasal cannula. His pulmonary function study shows a forced vital capacity (FVC) of 55%, carbon dioxide diffusing capacity (DLCO) of less than 45% predicted. The echocardiogram shows features suggestive of pulmonary hypertension.
What features on initial evaluation in this patient predict adverse outcomes and higher mortality? Answer: Pulmonary hypertension
The natural history of IPF is heterogeneous with certain patients having a rapid progression with recurrent exacerbations, but the disease may remain stable in a subset of patients for a number of years. In many instances, patients do not seek medical attention until the symptoms are overwhelming. In fact, delayed access to a tertiary care center was associated with higher mortality independent of risk factors.16 The prevalence is higher in elderly, male patients with history of smoking and usually follows a chronic progressive course.
Several scoring systems and biomarkers are being developed to identify patients at high risk of progression. The 2011 guidelines identify baseline and longitudinal risk factors associated with increased risk of progression and higher mortality in patients with IPF (Table 4). Biomarkers such as surfactant proteins A and D Krebs von den Lungen Antigen (KL-6), glycoprotein, chemokine CCL18 matrix metalloproteinases, and circulating fibrocytes, have all been shown to be elevated in
patients with IPF, though their use outside a research setting has not been well-documented.17
In 2012, a multidimensional model for prognosis and staging of IPF was developed as a simple prognostication tool for predicting mortality in patients with IPF.18 Named GAP Index for its use of gender, age and physiology as key predictive variables, a modified index was recently developed for application across all ILD subtypes to provide disease-specific survival estimates and to accurately predicts mortality among all chronic ILD (Table 5).19
There has been an increased recognition of a distinct clinical syndrome known as combined pulmonary fibrosis emphysema in the upper lobes and pulmonary fibrosis in the lower lobes in the same patient.20,21 Some patients may have a normal spirometry, however, some will have severe impairment in diffusion
capacity/gas exchange. Seen mostly in male smokers, CPFE is associated with higher mortality, pulmonary hypertension, acute lung injury, and lung cancer.
Patients with suspected pulmonary hypertension may undergo right heart catheterization for diagnosis, since echocardiographic features do not correlate well with right-sided pressures in IPF. The pathobiology is related with endothelial apoptosis and growth factor-induced remodeling of pulmonary arteries and not just hypoxemia-induced pulmonary vasoconstriction as previously thought.22 These patients have a higher mortality and are proportional to elevated pulmonary arterial pressures. Pulmonary hypertension in IPF patients is best correlated by gas exchange efficiency during exercise and peak oxygen uptake.23 The latest set of guidelines does not recommend treatment in the majority of IPF patients with pulmonary hypertension, as it may cause worsening hypoxemia. However, in a selected minority of patients, oral sildenafil has shown to improve exercise capacity.24
Patients with IPF are also at risk for other coexisting diseases such as GERD, venous thromboembolism, coronary artery disease, sleep-disordered breathing, depression, and lung cancer.25 Recent data suggests poor outcomes in patients with IPF and sleep apnea, compounded by nocturnal desaturation.26 There has also been increased awareness of recurrent microaspiration and asymptomatic gastroesophageal reflux disease (GERD) in patients with IPF.27 The 2011 guidelines recommend medically treating the majority of IPF patients with GERD. There is an increased prevalence of lung cancer in IPF patients and these patients have a higher mortality compared to those without.28
History: Patient returns for a routine follow-up visit. His symptoms have remained stable over the last 4 weeks. He is interested in pharmacological treatment options for his condition.
What are all the new FDA-approved treatment options currently available for IPF patients? Answer: Nintedanib and Pirenidone.
Previously, the pathogenesis of IPF has been associated with lung injury and inflammation, but earlier clinical trials targeting inflammation have been consistently negative. The current paradigm of IPF pathogenesis is now understood to be more aberrant wound healing.29 In genetically susceptible individuals, nonresolving injury to alveolar epithelial cells consequent to environmental exposures triggers an aberrant wound healing process, leading to activation of several pathways with release of cytokines and growth factors. These are: fibroblast recruitment, proliferation, and activation with accumulation of myofibroblast; vascular leak and extravascular coagulation; formation of provisional matrix and cross-linking.30 In IPF patients, dysregulation of this process and absence of apoptosis of these activated myofibroblasts, leads to progressive deposition of collagen, formation of fibroblastic foci, and lung remodeling.
Several molecules targeting various pathways of injury, inflammation, and repair (eg, epithelial cell injury, immune system dysregulation, fibroblast accumulation, myofibroblast differentiation, and extracellular matrix deposition and stiffening), have completed phase 2 or 3 clinical trials. The 2011 guidelines did not find any conclusive evidence to specifically give strong recommendations for treatment with any particular agents (Table 6).

Pirenidone is an antifibrotic and antioxidant agent that has shown positive signals in CAPACITY-1 and CAPACITY-2 trials (Clinical Studies Assessing Pirenidone in Idiopathic Pulmonary Fibrosis: Research of Efficacy and Safety Outcomes), but the data was insufficient for FDA approval. Its exact mechanism of action is unknown. The recently published ASCEND (Assessment of Pirenidone to Confirm Efficacy and Safety in Idiopathic Pulmonary Fibrosis) demonstrated, over a 52-week period, that patiarents on pirenidone, at a dose of 800 mg, 3 times a day (n=278), compared to placebo, had a significantly decreased percentage of their lung function deterioration. (Defined as absolute decline of 10 percentage points or more in the percentage of the predicted FVC). In addition, when combining the patients from the CAPACITY and ASCEND trials, there was anzimprovement in mortality.31 Patients on pirenidone also had better exercise capacity with longer 6-minute-walk distance and progression-free survival. The side effects were mainly nausea and skin reactions.
Nintedanib is a tyrosine kinase inhibitor that targets various growth factors, including platelet derived growth factor receptors a/b, fibroblast growth factor receptor receptors 1 to 3, and vascular endothelial growth factor receptors 1 to 3. Data from the INPULSIS Trials 1 and 2 were published along with the ASCEND trial. Patients on nintedanib at a dose of 150 mg 2 times a day had significant lower decline in lung function as shown by adjusted annual rate of change in FVC compared to placebo.32 The predominant side effects were diarrhea and liver function abnormalities in the nintedanib group. Based on these 2 trials, the FDA has approved the use of both these medications for patients with IPF.
History The patient was started on pirenidone treatment. In addition to pharmacotherapy, what other measures are indicated as per 2011 ATS guidelines on IPF? Answer: Pulmonary rehabilitation
Once the diagnosis is made, the physician should spend adequate time with the patient to explain the prognosis and assess the patient's preferences and values. Typically, patients diagnosed with IPF have a higher prevalence of depression.33 The burden of the disease is emotionally overwhelming for the patients and the ramification of having a morbid disease will likely impact family members as well. In our institution, we routinely enroll patients in pulmonary fibrosis support groups and offer psychological and emotional support to help cope with the disease. Patients who are at increased risk of mortality should be referred for lung transplantation early in the course. Most patients with IPF have exercise-induced hypoxemia. A 6-minute or shuttle-walk test will provide the most accurate indication of exercise-induced desaturation and the required level of oxygen supplementation.
Although there are no randomized controlled trials available in patients with IPF, ambulatory oxygen or long-term oxygen therapy in patients with exercise-induced hypoxemia and resting desaturation may improve exercise capacity and prevent development of pulmonary hypertension. The majority of patients who die with IPF are in the hospital setting, and the palliative team are consulted late in the disease.34 End-of-life management in IPF should be brought up earlier in patient discussion, and if appropriate, early referral to palliative care specialist should be done.
The majority of patients with IPF have some sort of exercise intolerance due to altered respiratory mechanics, impaired gas exchange, peripheral muscle dysfunction, and circulatory limitation. Pulmonary rehabilitation involves light aerobic exercises, endurance training, respiratory breathing techniques, and educational sessions offered by respiratory therapists, physiotherapists, and nurses—therapies that have been shown to improve the health-related quality of life, decrease dyspnea score, and improve exercise endurance. Even late in the disease, these interventions should be offered to all IPF patient.35,36
History The patient was enrolled in pulmonary rehabilitation program and IPF support group. 6 months later, the patient was admitted with severe dyspnea and hypoxemia. A repeat CT scan of the chest ruled out pulmonary embolism, but showed bilateral GGO in addition to previously noted fibrotic changes. A bronchoscopy was performed and cultures were negative. The echocardiogram and brain natriuretic peptide (BNP) were within normal range.
Which medication should be started at this point? Answer Corticosteroid.
Patients with diagnosed IPF are at risk of having acute respiratory decline due to common conditions such as pneumonia, pulmonary embolism, pneumothorax, or cardiac failure. Acute exacerbation of IPF by definition is a diagnosis of exclusion and is known to occur in 5% to 10% of patients yearly.37,38 It is unknown if this is an inherent acceleration in the pathobiological process. Patients present with worsening cough, fever, and dyspnea. Although no specific risk factors are identified, it is known to occur in patients after having thoracic surgery and bronchoalveolar lavage (BAL). HRCT shows GGO superimposed on UIP pattern. Increased levels of fibrocytes and alpha defensins are noted in serum and BAL in patients with acute exacerbation and pathology usually shows diffuse alveolar damage or organizing pneumonia.39 The treatment is supportive and majority of patients should be started on high dose–pulse corticosteroid treatment.5 The mortality related with acute exacerbation is high, though lung transplantation might be an option in selected patients. Unfortunately, most institutions are not equipped to offer (perform) transplants.
Conclusion
Pulmonary fibrosis poses a difficult challenge in diagnosis and treatment for the physician. Interstitial pulmonary fibrosis, in particular, can be difficult to diagnose and can be associated with a very poor prognosis. Until recently, treatment was largely supportive, including oxygen, pulmonary rehabilitation, and treatment of comorbid conditions. Now, however, 2 new FDA-approved medications have been shown to slow the progressive loss of lung function in this disease. Further research into other fibrotic pathways, combinations of anti-fibrotic agents, and the use of these agents in other fibrotic lung disease has the potential to greatly enhance our treatment strategies in these difficult disorders.
Girish B. Nair, MD, FACP, FCCP, is director of the Interstitial Lung Disease and Pulmonary Rehabilitation programs at Winthrop-University Hospital, as well as assistant professor of medicine, SUNY at Stony Brook in New York.
Jonathan Ilowite, MD, FCCP, is codirector of the Interstitial Lung Disease Program, associate division chief pulmonary and critical care, program director pulmonary fellowship program at Winthrop-University Hospital and associate professor department of medicine, SUNY at Stony Brook in New York.
References:
1.Travis WD, Costabel U, Hansell DM, et al. An official American Thoracic Society/European Respiratory Society statement: update of the international multidisciplinary classification of the idiopathic interstitial pneumonias. Am J Respir Crit Care Med. 2013;188(6):733-748.
2.Raghu G, Weycker D, Edelsberg J, et al. Incidence and prevalence of idiopathic pulmonary fibrosis. Am J Respir Crit Care Med. 2006;174(7):810-816.
3. Ryu JH, Olson EJ, Midthun DE, Swensen SJ. Diagnostic approach to the patient with diffuse lung disease. Mayo Clin Proc. 2002;77(11):1221-1227.
4. Palmucci S, Roccasalva F, Puglisi S, et al. Clinical and radiological features of idiopathic interstitial pneumonias (IIPs): a pictorial review. Insights Imaging. 2014;5(3):347-364
5.Raghu G, Collard HR, Egan JJ, et al. An official ATS/ERS/JRS/ALAT statement: idiopathic pulmonary fibrosis: evidence-based guidelines for diagnosis and management. Am J Respir Crit Care Med. 2011;183(6):788-824.
6.Flaherty KR, Toews GB, Travis WD, et al. Clinical significance of histological classification of idiopathic interstitial pneumonia. Eur Respir J. 2002;19(2):275-283.
7.Quadrelli S, Molinari L, Ciallella L, et al. Radiological versus histopathological diagnosis of usual interstitial pneumonia in the clinical practice: does it have any survival difference? Respiration. 2010;79(1):32-37.
8.Sumikawa H, Johkoh T, Colby T, et al. Computed tomography findings in pathological usual interstitial pneumonia: relationship to survival. Am J Respir Crit Care Med. 2008;177(4):433-439.
9.Oda K, Ishimoto H, Yatera K, et al. High-resolution CT scoring system-based grading scale predicts the clinical outcomes in patients with idiopathic pulmonary fibrosis. Respir Res. 2014;15:10.
10.Meyer KC, Raghu G, Baughman RP, et al. An official American Thoracic Society clinical practice guideline: the clinical utility of bronchoalveolar lavage cellular analysis in interstitial lung disease. Am J Respir Crit Care Med. 2012;185(9):1004-1014.
11.Flaherty KR, Thwaite EL, Kazerooni EA, et al. Radiological versus histological diagnosis in UIP and NSIP: survival implications. Thorax. 2003;58(2):143-148.
12.Monaghan H, Wells AU, Colby TV, et al. Prognostic implications of histologic patterns in multiple surgical lung biopsies from patients with idiopathic interstitial pneumonias. Chest. 2004;125(2):522-526.
13.Flaherty KR, Travis WD, Colby TV, et al. Histopathologic variability in usual and nonspecific interstitial pneumonias. Am J Respir Crit Care Med. 2001;164(9):1722-1727.
14.Thomeer M, Demedts M, Behr J, et al. Multidisciplinary interobserver agreement in the diagnosis of idiopathic pulmonary fibrosis. Eur Respir J. 2008;31(3):585-591.
15.Flaherty KR, King TE Jr, Raghu G, et al. Idiopathic interstitial pneumonia: what is the effect of a multidisciplinary approach to diagnosis? Am J Respir Crit Care Med. 2004;170(8):904-910.
16.Lamas DJ, Kawut SM, Bagiella E, et al. Delayed access and survival in idiopathic pulmonary fibrosis: a cohort study. Am J Respir Crit Care Med. 2011;184(7):842-847.
17.Cicchitto G, Sanguinetti CM. Idiopathic pulmonary fibrosis: the need for early diagnosis. Multidiscip Respir Med. 2013;8(1):53.
18.Ley B, Ryerson CJ, Vittinghoff E, et al. A multidimensional index and staging system for idiopathic pulmonary fibrosis. Ann Intern Med. 2012;156(10):684-691.
19.Ryerson CJ, Vittinghoff E, Ley B, et al. Predicting survival across chronic interstitial lung disease: the ILD-GAP model. Chest. 2014;145(4):723-728.
20.Cottin V, Nunes H, Brillet PY, et al. Combined pulmonary fibrosis and emphysema: a distinct underrecognised entity. Eur Respir J. 2005;26(4):586-593.
21.Jankowich MD, Rounds SI. Combined pulmonary fibrosis and emphysema syndrome: a review. Chest. 2012;141(1):222-231.
22.Farkas L, Gauldie J, Voelkel NF, Kolb M. Pulmonary hypertension and idiopathic pulmonary fibrosis: a tale of angiogenesis, apoptosis, and growth factors. Am J Respir Cell Mol Biol. 2011;45(1):1-15.
23.Glaser S, Obst A, Koch B, et al. Pulmonary hypertension in patients with idiopathic pulmonary fibrosis-the predictive value of exercise capacity and gas exchange efficiency. PLoS One. 2013;8(6):e65643.
24.Collard HR, Anstrom KJ, Schwarz MI, Zisman DA. Sildenafil improves walk distance in idiopathic pulmonary fibrosis. Chest. 2007;131(3):897-899.
25.King C, Nathan SD. Identification and treatment of comorbidities in idiopathic pulmonary fibrosis and other fibrotic lung diseases. Curr Opin Pulm Med. 2013;19(5):466-473.
26.Kolilekas L, Manali E, Vlami KA, et al. Sleep oxygen desaturation predicts survival in idiopathic pulmonary fibrosis. J Clin Sleep Med. 2013;9(6):593-601.
27.Lee JS, Ryu JH, Elicker BM, et al. Gastroesophageal reflux therapy is associated with longer survival in patients with idiopathic pulmonary fibrosis. Am J Respir Crit Care Med. 2011;184(12):1390-1394.
28.Tomassetti S, Gurioli C, Ryu J, et al. The impact of lung cancer on survival of Idiopathic Pulmonary Fibrosis. Chest. 2015;147(1):157-164.
29.Ahluwalia N, Shea BS, Tager AM. New therapeutic targets in idiopathic pulmonary fibrosis. Aiming to rein in runaway wound-healing responses. Am J Respir Crit Care Med. 2014;190(8):867-878.
30.King TE Jr, Pardo A, Selman M. Idiopathic pulmonary fibrosis. Lancet. 2011;378(9807):1949-1961.
31.King TE Jr, Bradford WZ, Castro-Bernardini S, et al. A phase 3 trial of pirenidone in patients with idiopathic pulmonary fibrosis. N Engl J Med. 2014;370(22):2083-2092.
32.Richeldi L, du Bois RM, Raghu G, et al. Efficacy and safety of nintedanib in idiopathic pulmonary fibrosis. N Engl J Med. 2014;370(22):2071-2082.
33.Akhtar AA, Ali MA, Smith RP. Depression in patients with idiopathic pulmonary fibrosis. Chron Respir Dis. 2013;10(3):127-133.
34.Lindell KO, Liang Z, Hoffman LA, et al. Palliative Care and Location of Death in Decedents with Idiopathic Pulmonary Fibrosis. Chest. 2014.
35.Holland AE, Hill CJ, Conron M, et al. Short term improvement in exercise capacity and symptoms following exercise training in interstitial lung disease. Thorax. 2008;63(6):549-554.
36.Spruit MA, Singh SJ, Garvey C, et al. An official American Thoracic Society/European Respiratory Society statement: key concepts and advances in pulmonary rehabilitation. Am J Respir Crit Care Med. 2013;188(8):e13-64.
37.Martinez FJ, Safrin S, Weycker D, et al. The clinical course of patients with idiopathic pulmonary fibrosis. Ann Intern Med. 2005;142(12 Pt 1):963-967.
38.Ambrosini V, Cancellieri A, Chilosi M, et al. Acute exacerbation of idiopathic pulmonary fibrosis: report of a series. Eur Respir J. 2003;22(5):821-826.
39.Bhatti H, Girdhar A, Usman F, et al. Approach to acute exacerbation of idiopathic pulmonary fibrosis. Ann Thorac Med. 2013;8(2):71-77.