
Periorbital Edema and Abdominal Distension: Beyond Celiac Disease
A 17½-month-old boy presented to the pediatric gastroenterology office with a 5-month history of emesis and 4 large, pale, and frothy-appearing bowel movements a day. During a visit to his primary care provider when he was 13 months of age, the child was found to have had a normocytic anemia (hemoglobin, 10.1 g/dL; hematocrit, 29%; mean corpuscular volume, 85 µm3) and hypoalbuminemia (albumin, 2.3 g/dL; total serum protein, 4.7 g/dL), with a normal iron level (45 µg/dL) and a low total iron-binding capacity (199 µg/dL).
At the time of presentation to the pediatric gastroenterology office, he also had developed abdominal distension and intermittent periorbital edema. His length had been in the 50th percentile at 9 months of age but now was below the 5th percentile. His weight had plateaued at 15 months of age, and he had a normal weight-to-height ratio (Figure 1).

Figure 1. Growth chart demonstrating linear growth failure and poor weight gain by 17½ months of age.
His tissue transglutaminase IgA antibody (tTG-IgA) level was 13 U/mL; his deamidated gliadin IgA and IgG antibody levels were 139.2 U and 13.1 U, respectively, with normal levels of total IgA (227 mg/dL) and prealbumin (30 mg/dL). The C-reactive protein level was 0.3 mg/L, and the erythrocyte sedimentation rate was 51 mm/h.
Urinalysis revealed 2+ protein and microscopic hematuria on first morning urine sample. Fecal α1-antitrypsin levels were normal at 0.17 mg/g. There is a family history of ulcerative colitis in his immediate family, as well as diabetes and hypothyroidism in other relatives.
Upper endoscopy and flexible sigmoidoscopy results were significant for duodenal mucosal scalloping (Figure 2) and solid stool in the rectum. Biopsy results revealed Marsh stage 3C duodenitis (total villous atrophy) (Figure 3) and normal esophageal, gastric, and distal colonic mucosae.

Figure 2. Endoscopy of the second portion of the patient’s duodenum revealing visual scalloping of the mucosa.
He was started on a gluten-free diet. In response to his nephrotic syndrome, treatment with 2 mg/kg/d of prednisone was initiated for possible minimal change disease, given negative test results for antinuclear antibodies, antineutrophil cytoplasmic antibodies (cytoplasmic and perinuclear), C3, C4, antiglomerular basement membrane antibodies, and anti–double-stranded DNA antibodies.
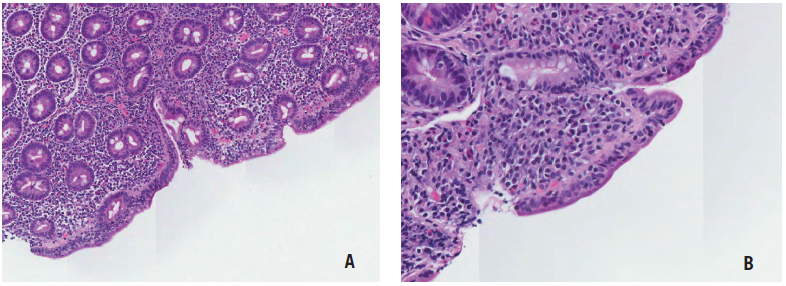
Figure 3. Hematoxylin and eosin (H&E) stain of the duodenum biopsy sample at ×80 magnification (A) and ×200 magnification (B) revealing increased intraepithelial lymphocytes and villous atrophy.
Although his proteinuria did not improve, his serum albumin level improved to 3.8 g/dL, and his diarrhea, vomiting, and edema resolved, as did his anemia and hypoalbuminemia. Given his short stature, levels of insulinlike growth factor I and insulinlike growth factor binding protein 3 were obtained; results were normal at 108 ng/mL and 2.6 mg/L, respectively.

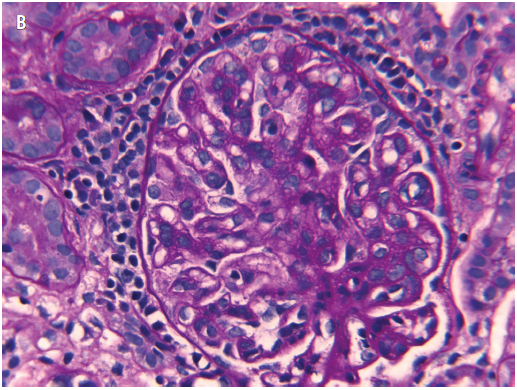
Figure 4. A, H&E stain of a representative field of the renal biopsy sample, with a glomerulus showing an increase in mesangial and endocapillary cells. B, Periodic acid-Schiff stain of the renal biopsy sample highlighting excess intercapillary amorphous matrix and thickened basement membranes.
Results of a renal biopsy, performed to evaluate the etiology of his nephrotic syndrome that was unresponsive to corticosteroids, revealed IgA nephropathy (Figures 4-6). He continued to have significant proteinuria, despite prednisone therapy and the clinical resolution of his celiac enteropathy.
At 21 months of age, his short stature had resolved on the gluten-free diet. Follow-up deamidated gliadin antibody levels were obtained to evaluate for dietary adherence as a possible co-contributor to his persistent renal disease; results were negative for IgA (2 U) and IgG (6 U).
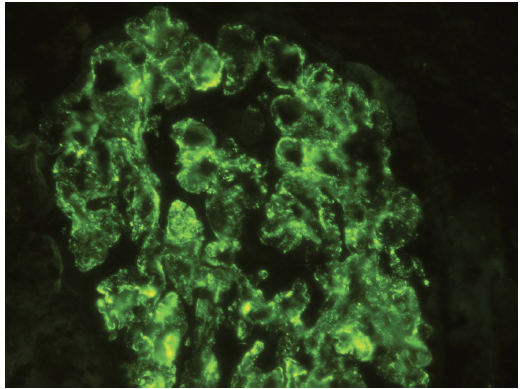
Figure 5. Immunofluorescence stain for IgA demonstrating diffuse, glomerular granular staining (3+ to 4+ on a 0 to 4+ scale). Weak staining for IgG, IgM, C3, and C4 (not represented) also was present.
Discussion
Celiac disease (CD) results from an autoimmune process in response to gliadin stimulation in the proximal small intestine, with the development of specific antibodies to the gliadin protein in wheat and to intestinal cell structural proteins in the connective tissue (transglutaminase antibodies and anti-endomysial antibodies). As much as 1% of the population of Western Europe and North America have CD.1 CD is associated with many other immunologic disorders, such as diabetes mellitus, thyroiditis, chronic active hepatitis, IgA deficiency, Sjögren syndrome, idiopathic pulmonary fibrosis, cow’s milk protein-sensitive enteropathy, and IgA nephropathy, as well as dermatitis herpetiformis, Down syndrome, cystic fibrosis, and α1-antitrypsin deficiency.
IgA nephropathy is the most common form of glomerular nephritis associated with CD.2 In one study, Ludvigsson and colleagues1 found that the overall risk of renal disease due to any glomerular nephritis was higher among patients with CD (hazard ratio, 1.64; 95% confidence interval, 1.01-2.66; P = .046), corresponding to 0.2% of CD patients compared with 0.1% of the general population, with a median age at first diagnosis of 28 years (range, 4-86 years).
It has been reported that there is no HLA-DQ haplotype-related risk of celiac disease in patients with IgA nephropathy.2,3 While hypoalbuminemia is a well-known potential consequence of CD due to the associated protein-losing enteropathy, nephrotic syndrome is rarely considered a contributing factor to hypoalbuminemia in CD. A scarcity of literature exists about pediatric renal disease related to CD. A literature search yields only one case report of a 12-year-old girl in Turkey with CD and membranoproliferative glomerulonephritis,4 and one letter to the editor stating that 5 of 251 pediatric patients with nephrotic syndrome seen in a hospital in Barcelona, Spain, also had CD5; the letter-writers note that kidney biopsies were done in only 2 of the 5 patients, one of which showed minimal change disease lesions and the other of which showed IgM infiltrates.
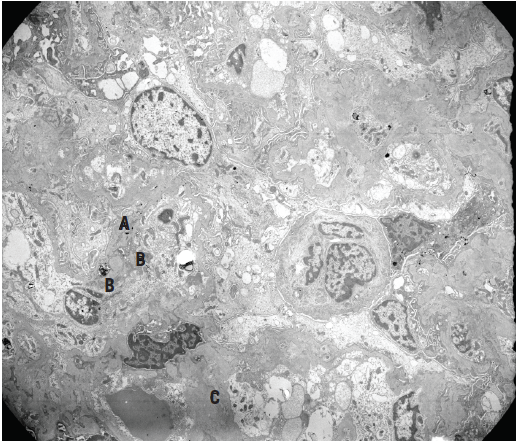
Figure 6. Electron micrograph of a glomerular tuft showing irregularly thickened peripheral capillary basement membranes with scattered subepithelial (A), subendothelial (B), and intramembranous dense deposits, as well as abundant mesangial and paramesangial deposits (C), corresponding to IgA deposition.
To our knowledge, concurrent nephrotic syndrome identified at the time of diagnosis of CD in a patient younger than 3 years of age has not been previously reported. Despite the expectation that the patient’s hypoalbuminemia and intermittent edema would prove to be a result of CD involvement of the gastrointestinal tract, instead the patient’s kidney disease was the culprit. There was no evidence of a protein-losing enteropathy, based on the patient’s normal fecal α1-antitrypsin level with documented proteinuria.
Periorbital edema and abdominal distension in toddlers should lead to the consideration of and serology assessment for CD, as well as for other possible etiologies of hypoalbuminemia, such as nephrotic syndrome. All instances of hypoalbuminemia in CD may not be a result of a protein-losing enteropathy secondary to the duodenitis or enteritis. Although associated nephrotic syndrome is uncommon in patients with CD, awareness of this pathology is necessary when assessing and treating them, and a urinalysis should be done in patients with CD and hypoalbuminemia to evaluate for this comorbid renal disorder.
References:
1. Ludvigsson JF, Montgomery SM, Olén O, Ekbom A, Ludvigsson J, Fored M. Coeliac disease and risk of renal disease—a general population cohort study. Nephrol Dial Transplant. 2006;21(7):1809-1815.
2. Collin P, Syrjänen, J, Partanen J, Pasternack A, Kaukinen K, Mustonen J. Celiac disease and HLA DQ in patients with IgA nephropathy. Am J Gastroenterol. 2002;97(10):2572-2576.
3. Mignot E, Kimura A, Abbal N, et al. DQCAR microsatellite polymorphisms in three selected class II-associated diseases. Tissue Antigens. 1995;46:299-304.
4. Bıyıklı NK, Gökçe I, Çakalağoğlu F, Arbak S, Alpay H. The co-existence of membranoproliferatıve glomerulonephritis type 1 and coeliac disease: a case report. Pediatr Nephrol. 2009;24(6):1247-1250.
5. Gimenez Llort A, Vila Cots J, Camacho Diaz JA, Vila Santandreu A, Concheiro Guisan A, Garcia Garcia L. Nephrotic syndrome associated with celiac disease: a report of five cases. Nephron. 2002;92(4):950.
Dr Kader is an assistant professor and Ms Martin is a staff nurse, both in the Department of Pediatrics, Division of Gastroenterology and Nutrition; Dr Twaddell is an assistant professor and Dr Drachenberg is a professor, both in the Department of Pathology; and Dr Jinadu is an assistant professor in the Department of Pediatrics, Division of Nephrology, all at the University of Maryland School of Medicine in Baltimore.