
Peer Reviewed
Ectopic ACTH Syndrome From Metastatic Pancreatic Neuroendocrine Tumor
Authors:
Richa Handa, MD, and Akhil Rahman, MD
Citation:
Handa R, Rahman A. Ectopic ACTH syndrome from metastatic pancreatic neuroendocrine tumor. Consultant. 2017;57(8):472-474.
Ectopic adrenocorticotropic hormone (ACTH) syndrome is a rare disease with a reported incidence of 0.1 cases per million per year.1,2 Various cancers can cause ectopic ACTH syndrome.1,2 Hypercortisolism (Cushing syndrome) and metabolic changes such as hypokalemia (which is found in almost 70% of cases) and metabolic alkalosis are common in patients with ectopic ACTH syndrome.3
Pancreatic neuroendocrine tumors are rare neoplasms with an incidence of 1 case per 100,000 per year. Chromogranin A (CgA) and synaptophysin test results are positive in patients with these tumors.4-7 Pancreatic neuroendocrine tumors are also associated with various genetic syndromes such as multiple endocrine neoplasia type 1, von Hippel-Lindau disease, neurofibromatosis, and tuberous sclerosis. Functioning pancreatic neuroendocrine tumors cause clinical syndromes due to hypersecretion of various hormones.
Patients with an ACTH-secreting pancreatic neuroendocrine tumor usually present with liver metastasis, because ACTH released by a pancreatic tumor enters enterohepatic circulation, is rapidly metabolized by the liver, and does not produce any symptoms, whereas ACTH produced by bronchial, ovarian, and other neuroendocrine tumors enters the systemic circulation and results in clinical manifestations earlier, leading to earlier diagnosis.8,9 In one case series, 10 patients with hypercortisolism secondary to ectopic ACTH secretion by a pancreatic neuroendocrine tumor were studied; all of them showed liver metastases at presentation.10 A review of the known 42 cases of pancreatic neuroendocrine tumors associated with hypercortisolism found 88% of patients to have metastases, 60% of whom died in 2 years or less.11
We present a case in which a patient presenting with diarrhea eventually received a diagnosis of metastatic neuroendocrine tumor involving the liver, with the primary tumor located in the pancreas.
CASE PRESENTATION
A 51-year-old man with a history of hypertension, insulin-dependent diabetes mellitus, and morbid obesity after gastric bypass surgery presented to the emergency department with a 4-week history of diarrhea, generalized weakness, weight gain, and lower extremity swelling. He had also noticed high blood glucose levels despite having used the prescribed insulin dosage.
Physical examination revealed elevated blood pressure at 182/65 mm Hg, obesity, abdominal distension, and bilateral lower extremity pitting edema. There were no other focal signs.
Results of initial laboratory studies showed severe hypokalemia with a potassium level of less than 1.5 mEq/L (reference range, 3.5-5.0 mEq/L), metabolic alkalosis with a carbon dioxide level of 40 mEq/L (reference range, 22-31 mEq/L), and hypochloremia with a chloride level of 92 mEq/L (reference range, 98-108 mEq/L). The patient’s renal function was within normal range. The albumin level was low at 2.3 g/dL (reference range, 3.5-5.0 g/dL).
The patient’s liver function test results were also abnormal, with an alkaline phosphate level of 249 U/L (reference range, 26-126 U/L), an aspartate aminotransferase level of 74 U/L (reference range, 0-46 U/L), and an alanine aminotransferase level of 69 U/L (reference range, 6-40 U/L). Cortisol levels were also elevated at 101.4 µg/dL (reference range, 5-25 µg/dL) and remained high even after a high-dose dexamethasone suppression test. ACTH levels were elevated at 164 pg/mL (reference range, < 46 pg/mL). Stool test results showed no ova or parasites but were positive for a fat level of more than 100 drops/hpf, and the results of a celiac disease panel were negative.
Computed tomography (CT) scans of the chest and abdomen/pelvis showed innumerable low-attenuation lesions scattered throughout the liver, with the largest measuring up to 5.5 cm in diameter (Figures 1A-1C); the lesions were likely related to metastasis. A questionable lesion was also noticed at the head of the pancreas (Figure 2), and the scan showed bowel-wall thickening involving the proximal descending colon (Figure 3).
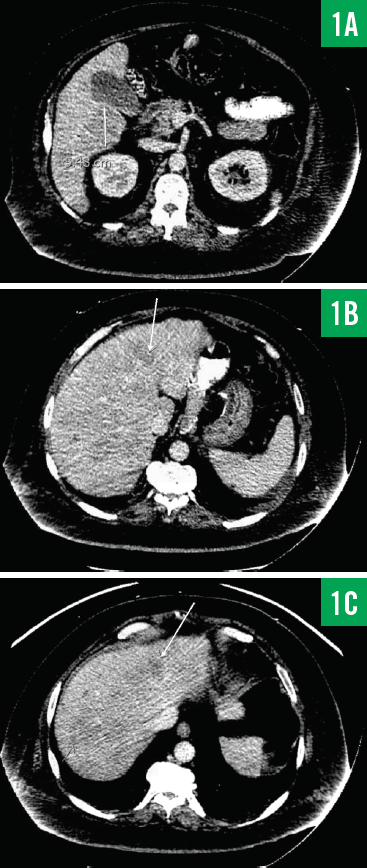
Figure 1. CT scans of the patient’s abdomen/pelvis showing a 5.48-cm lesion involving the inferior aspect of the right lower lobe of the liver (1A) and additional smaller lesions involving the liver (1B and 1C).
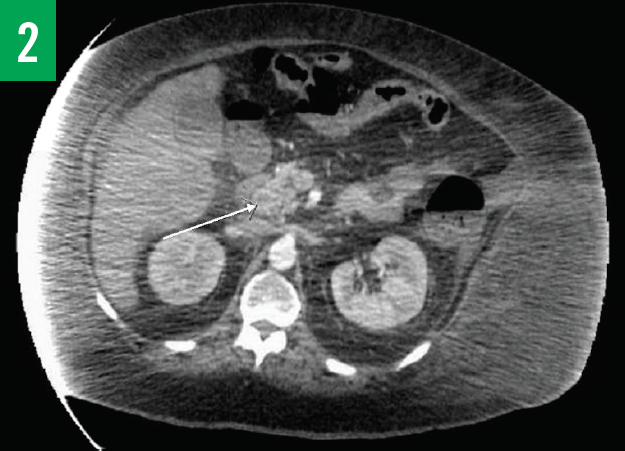
Figure 2. CT scan with contrast of the chest showing a questionable lesion at the head of pancreas.
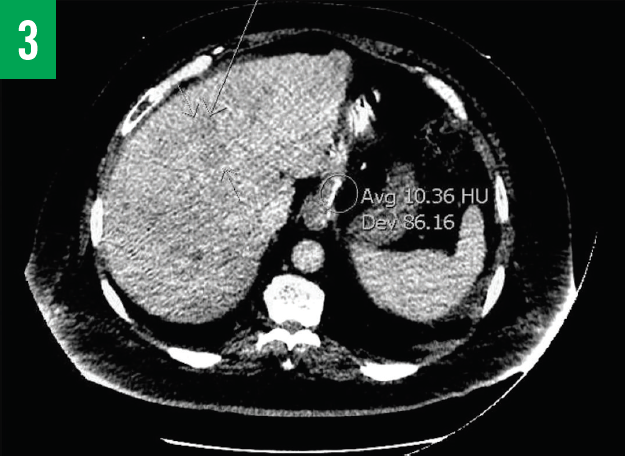
Figure 3. CT scan of the abdomen/pelvis showing numerous low-attenuation lesions scattered throughout the liver and bowel thickening involving the proximal part of the descending colon.
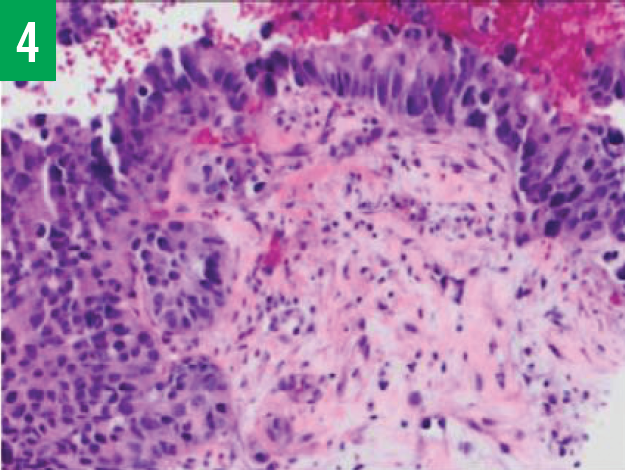
Figure 4. Histology test results of liver biopsy specimens showing a nested group of neuroendocrine cells with moderate cytoplasm and large nuclei.
The patient underwent colonoscopy, the findings of which were within normal limits. Esophagogastroduodenoscopy with biopsy was also done, the results of which showed ulceration at the gastroenteric anastomosis, but the biopsy results showed no evidence of malignancy. Because of the presence of multiple low-attenuation lesions, the patient also underwent CT-guided liver biopsies, the histology results of which were positive for poorly differentiated large-cell neuroendocrine carcinoma (Figure 4). Liver biopsies were also positive for the neuroendocrine markers CgA, synaptophysin, neuron-specific enolase, and CD56. Cancer antigen 19-9 levels were very high at 2027.9 U/mL (reference range, ≤ 35 U/mL). Carcinoembryonic antigen levels were elevated at 17.7 ng/mL (reference range, 0-5.0 ng/mL).
During the course of hospitalization, the patient remained hypertensive and hypokalemic even after regular potassium replacement. Eventually, the patient was started on chemotherapy because of the extensive liver metastases. Despite the aggressive treatment, the patient died after 3 weeks of hospitalization.
DISCUSSION
The presentation of ectopic ACTH production secondary to a pancreatic neuroendocrine tumor can be very vague and can vary from diarrhea (as in our patient’s case) to life-threatening manifestations such as hypertensive crisis and diabetic ketoacidosis.12 Pancreatic neuroendocrine tumors usually present with advanced and metastatic disease, which is found in almost 75% of cases.13,14 These tumors also have a high recurrence rate, despite resection of the primary lesion, and their secretion of multiple hormones leads to differing clinical manifestations.15
Finding the source of ectopic ACTH production sometimes can be difficult because of the small size of the tumor. Approximately 12% to 19% of tumors are not identified despite extensive workup.16 In our patient’s case, we were able to see multiple small lesions on the liver during imaging, which helped us diagnose the condition. Primary sites usually include the lungs, but cases have been reported in which the primary site has been the pancreas (8%), thymus (11%), mid gut, hind gut, prostate, ovary, uterus, cervix, and thyroid.16-18 Approximately two-thirds of the tumors are located in the neck, thorax, and adrenal glands, and only one-third are found in intra-abdominal organs.19
The combination of octreotide scan, CT, magnetic resonance imaging, and positron emission tomography scans have been used to localize the tumor.20-22 Bilateral inferior petrosal sinus sampling usually shows absent central to peripheral gradient and is considered to be the gold standard for differential diagnosis.16 In our patient’s case, biochemical tests, imaging, immune cytochemical studies, and cytological studies helped us to diagnose ectopic ACTH syndrome.
Treatment usually involves medical and surgical therapy, given that these tumors have a very high recurrence rate of 30% to 50%.18 Dopamine agonists, ketoconazole, mitotane, metyrapone, etomidate, and aminoglutethimide have been used to control steroidogenesis.23,24 More specific treatment with somatostatin analogues such as octreotide, lanreotide, and pasireotide has also been used.24 Interferon-alfa, which inhibits angiogenesis, stimulates T cells, and induces cell-cycle arrest, also has been used.24 Various chemotherapeutic agents such as everolimus (an inhibitor of the mammalian target of rapamycin)25 and sunitinib and bevacizumab (angiogenesis inhibitors)26 have also been used. Combination therapy with streptozocin-doxorubicin or streptozocin-fluorouracil has been used with limited success.27,28
Ultimately, palliative care including arterial embolization and radiofrequency ablation of unresectable tumors is the goal of treatment in some patients.13 Debulking can also be performed to reduce tumor load. Our patient had received a diagnosis of a poorly differentiated large-cell neuroendocrine tumor and had positive test results for CgA and synaptophysin. The patient did not undergo surgical resection because of extensive metastases involving the liver, but he did undergo chemotherapy before his death after 3 weeks of hospitalization.
CONCLUSION
Pancreatic neuroendocrine tumors have variable but sometimes life-threatening manifestations. A high index of suspicion and early diagnosis are important to decrease overall mortality. The diagnosis of neuroendocrine tumors is usually delayed by 7 years from the first symptoms.29 Prognosis depends mainly on the histology of primary tumor. The 5-year survival rate of patients with pancreatic neuroendocrine tumor is 65%, and the 10-year survival rate is 45%, depending on the stage and grade of the tumor, whereas the survival rate of patients with pancreatic neuroendocrine carcinoma ranges from 1 month to 1 year.30
Richa Handa, MD, is a resident in the Internal Medicine Residency Program at Henry Ford Allegiance Health in Jackson, Michigan.
Akhil Rahman, MD, is an internal medicine physician at Henry Ford Allegiance Health in Jackson, Michigan.
REFERENCES:
- Isidori AM, Lenzi A. Ectopic ACTH syndrome. Arq Bras Endocrinol Metab. 2007;51(8):1217-1225.
- Baylin SB, Mendelsohn G. Ectopic (inappropriate) hormone production by tumors: mechanisms involved and the biological and clinical implications. Endocr Rev. 1980;1(1):45-77.
- Aron DC, Findling JW, Tyrrell JB. Glucocorticoids and adrenal androgens. In: Gardner DG, Shoback D, eds. Greenspan’s Basic and Clinical Endocrinology. 8th ed. New York, NY: McGraw-Hill; 2007:346-395.
- Strosberg JR, Cheema A, Weber J, Han G, Coppola D, Kvols LK. Prognostic validity of a novel American Joint Committee on Cancer staging classification for pancreatic neuroendocrine tumors. J Clin Oncol. 2011;29(22):3044-3049.
- Strosberg JR, Nasir A, Hodul P, Kvols L. Biology and treatment of metastatic gastrointestinal neuroendocrine tumors. Gastroinstest Cancer Res. 2008;2(3):113-125.
- Strosberg JR, Cheema A, Kvols LK. A review of systemic and liver-directed therapies for metastatic neuroendocrine tumors of the gastroenteropancreatic tract. Cancer Control. 2011;18(2):127-137.
- Milan SA, Yeo CJ. Neuroendocrine tumors of the pancreas. Curr Opin Oncol. 2012;24(1):46-55.
- Rindi G, Arnold R, Bosman FT, et al. Nomenclature and classification of neuroendocrine neoplasms of the digestive system. In: Bosman TF, Carneiro F, Hruban RH, Theise ND, eds. WHO Classification of Tumours of the Digestive System. Vol 3. 4th ed. Lyon, France: International Agency for Research on Cancer Press; 2010:13.
- Falconi M, Bartsch DK, Eriksson B, et al; Barcelona Consensus Conference Participants. ENETS consensus guidelines for the management of patients with digestive neuroendocrine neoplasms of the digestive system: well-differentiated pancreatic non-functioning tumors. Neuroendocrinology. 2012;95(2):120-134.
- Doppman JL, Nieman LK, Cutler GB Jr, et al. Adrenocorticotropic hormone-secreting islet cells tumors: are they always malignant? Radiology. 1994;190(1):59-64.
- Clark ES, Carney JA. Pancreatic islet cell tumor associated with Cushing’s syndrome. Am J Surg Pathol. 1984;8(12):917-924.
- Uecker JM, Janzow MT. A case of Cushing syndrome secondary to ectopic adrenocorticotropic hormone producing carcinoid of the duodenum. Am Surg. 2005;71(5):445-446.
- Steinmüller T, Kianmanesh R, Falconi M, et al. Consensus guidelines for the management of patients with liver metastases from digestive (neuro)endocrine tumors: foregut, midgut, hindgut, and unknown primary. Neuroendocrinology. 2008;87(1):47-62.
- Rodrigues P, Castedo JL, Damasceno M, Carvalho D. Ectopic Cushing’s syndrome caused by a pulmonary ACTH-secreting tumor in a patient treated with octreotide. Arq Bras Endocrinol Metabol. 2012;56(7):461-464.
- Solomou S, Khan R, Propper D, Berney D, Druce M. A case of insulin and ACTH co-secretion by a neuroendocrine tumour. Endocrinol Diabetes Metab Case Rep. 2014;2014:130082.
- Fasshauer M, Lincke T, Witzigmann H, et al. Ectopic Cushing’ syndrome caused by a neuroendocrine carcinoma of the mesentery. BMC Cancer. 2006;6:108.
- Sookur PA, Sahdev A, Rockall AG, et al. Imaging in covert ectopic ACTH secretion: a CT pictorial review. Eur Radiol. 2009;19(5):1069-1078.
- Alexandraki KI, Grossman AB. The ectopic ACTH syndrome. Rev Endocr Metab Disord. 2010;11(2):117-126.
- Ilias I, Torpy DJ, Pacak N, Mullen N, Wesley RA, Nieman LK. Cushing’s syndrome due to ectopic corticotropin secretion: twenty years’ experience at the National Institutes of Health. J Clin Endocrinol Metab. 2005;90(8):4955-4962.
- Özkan ZG, Kuyumcu S, Balköse D, et al. The value of somatostatin receptor imaging with In-111 octreotide and/or Ga-68 DOTATATE in localizing ectopic ACTH producing tumors. Mol Imaging Radionucl Ther. 2013;22(2):49-55.
- Bluemel C, Lapa C, Mottok A, Hahner S, Herrmann K. Tumor localization in ectopic Cushing syndrome using combined PET/CT imaging. Clin Nucl Med. 2013;38(9):749-751.
- Treglia G, Salomone E, Petrone G, Giaccari A, Rindi G, Rufini V. A rare case of ectopic adrenocorticotropic hormone syndrome caused by a metastatic neuroendocrine tumor of the pancreas detected by 68Ga-DOTANOC and 18F-FDG PET/CT. Clin Nucl Med. 2013;38(7):e306-e308.
- Sakihara S, Kageyama K, Yamagata S, Terui K, Daimon M, Suda T. A case of ectopic ACTH syndrome treated with intermittent administration of dopamine agonists. Endocrinol Diabetes Metab Case Rep. 2014;2014:140001.
- Fazio N, Cinieri S, Lorizzo K, et al. Biological targeted therapies in patients with advanced enteropancreatic neuroendocrine carcinomas. Cancer Treat Rev. 2010;36(suppl 3):S87-S94.
- Yao JC, Shah MH, Ito T, et al; RAD001 in Advanced Neuroendocrine Tumors, Third Trial (RADIANT-3) Study Group. Everolimus for advanced pancreatic neuroendocrine tumors. N Engl J Med. 2011;364(6):514-523.
- Raymond E, Dahan L, Raoul J-L, et al. Sunitinib malate for the treatment of pancreatic neuroendocrine tumors. N Engl J Med. 2011;364(6):501-513.
- Kouvaraki MA, Ajani JA, Hoff P, et al. Fluorouracil, doxorubicin, and streptozocin in the treatment of patients with locally advanced and metastatic pancreatic endocrine carcinomas. J Clin Oncol. 2004;22(23):4762-4771.
- Moertel CG, Lefkopoulo M, Lipsitz S, Hahn RG, Klaassen D. Streptozocin–doxorubicin, streptozocin–fluorouracil, or chlorozotocin in the treatment of advanced islet-cell carcinoma. N Engl J Med. 1992;326(8):519-523.
- Díez M, Teulé A, Salazar R. Gastroenteropancreatic neuroendocrine tumors: diagnosis and treatment. Ann Gastroenterol. 2013;26(1):29-36.
- Klimstra DS, Arnold R, Capella C, et al. Neuroendocrine neoplasms of the pancreas. In: Bosman TF, Carneiro F, Hruban RH, Theise ND, eds. WHO Classification of Tumours of the Digestive System. Vol 3. 4th ed. Lyon, France: International Agency for Research on Cancer (IARC) Press; 2010:322-326.