
Becker Muscular Dystrophy: Diagnosis and Lifelong Management
ABSTRACT: Although Duchenne muscular dystrophy (DMD) and Becker muscular dystrophy (BMD) result from mutations in the same gene and share many characteristics, BMD generally follows a milder and more protracted course. Early definitive diagnosis of BMD based on a thorough history and physical examination, laboratory testing, and genetic testing is critical to improving survival and preserving functional status. While there is no cure for BMD, supportive care from a multidisciplinary health care team can improve the quality of life of persons with BMD.
A 12-year-old boy presented to the clinic after his school bus driver noticed that the boy had been having difficulty climbing onto the bus. The boy’s difficulty had progressed to the point that he was seen crawling up the bus stairs. He also was reported to have been falling often at school lately, and he was having progressive difficulty with walking.
He reported no recent illnesses, difficulty breathing, or any other health concerns. At age 4 years, he had been diagnosed with mild pulmonary valve stenosis and since then had been following up with a cardiologist. His past medical history was significant for childhood asthma. He had no past surgical history.
He was born via normal, spontaneous, vaginal delivery, with no reported perinatal or neonatal problems. His parents, who are not consanguineous, and his five sisters were all healthy and lived in a mobile home with one pet dog.
The boy had experienced gross motor and speech developmental milestone delays, and at the time of presentation, he was at a functional age of 5 to 6 years. He had repeated kindergarten, and he had been receiving occupational therapy, physical therapy, and speech therapy at school. His immunizations were up to date.

Figure 1. Lower-extremity examination results included bilateral pseudohypertrophy of the calf muscles.
Physical Examination
On physical examination, the boy weighed 35 kg, placing him in the 90th percentile. Temperature was 36.4°C, his heart rate was 106 beats per minute, and his blood pressure was 100/60 mm Hg. He was sitting on an examination room table without any acute distress. No gross deformity or dysmorphism was visible. He had no craniofacial deformity, and his facial movements were normal and symmetric. His neck was supple, without any masses. Lungs were clear to auscultation, and air entry was good and even.
Results of the cardiovascular system examination included normal S1 and S2 sounds and a harsh grade 2 systolic murmur in the left upper sternal border. All peripheral pulses were equal bilaterally.
His abdomen was soft, nontender, and nondistended, with no organomegaly. He had Tanner stage I external male genitalia, with testes descended bilaterally.
Neurologically, he was alert, interactive, and very cooperative, with good eye contact and good social skills. Cranial nerves II through XII were grossly intact. Examination of the extremities revealed bilateral pseudohypertrophy of the calf muscles (Figure 1). He had no abnormal movements or posturing. Cerebellar signs were normal, and he had no ataxia.
Hypotonia was noted bilaterally in the upper and lower extremities, along with diffuse weakness of the proximal muscles as evidenced by a positive Gowers sign, which is elicited when a patient, in an attempt to stand from a supine or sitting position, rolls into the prone position, kneels, and pushes himself upright with his hands on the knees and thighs (Figure 2). Hyperlordosis was present; he had a waddling gait that at times was uncoordinated and awkward, and he walked on his tiptoes (Figure 3).

Figure 2. The boy used the Gowers maneuver to stand: While sitting (A), he rolled prone; knelt (B); and, with his hands on the floor (C), then his thighs, pushed himself upright (D).
Diagnostic Evaluation
The patient was referred to pediatric multispecialty care. The results of a muscle biopsy and serum genetic testing were suggestive of Becker muscular dystrophy (BMD). His serum creatine kinase (CK) level was elevated.
He was started on a regimen of prednisone, which led to his developing Cushing syndrome as an adverse effect of corticosteroid therapy. The results of a bone mineral density scan were abnormal, and he was started on a regimen of vitamin D and calcium.
Now at the age of 14, the boy has developed ankle contractures and has undergone bilateral Achilles-lengthening surgery. He also has developed kyphoscoliosis. He has become unable to do the Gowers maneuver to stand, because he can no longer use his thigh muscles. His gait has worsened, and he occasionally requires a wheelchair for mobility.

Figure 3. Physical examination showed hyperlordosis, pseudohypertrophy of the gastrocnemius muscle, and tiptoe posture upon standing.
Discussion
Dystrophinopathies are X-linked disorders (Figure 4) and are the most common inherited neuromuscular disease. Duchenne muscular dystrophy (DMD) is the most common variant, with an annual incidence of approximately 1 case per 3,600 male births,1 while its allelic variant, BMD, has an annual incidence of approximately 3 to 6 cases per 100,000 male births.2 Both dystrophinopathies result from a mutation in the gene encoding the protein dystrophin at the Xp21 locus. DMD features an almost complete absence of dystrophin production in muscle cells, whereas persons with BMD have partially functioning dystrophin.3 BMD was first described by Becker and Kiener in 19554,5 and is a separate clinical entity that generally follows a milder and more protracted course compared with DMD, despite the similarities (Table).1,3,6,7
All dystrophinopathies have X-linked recessive inheritance, but 30% of persons with dystrophinopathies have new mutations, and the mother is not a carrier. Female carriers are asymptomatic, but occasionally, affected girls and women can show much milder weakness compared with boys.
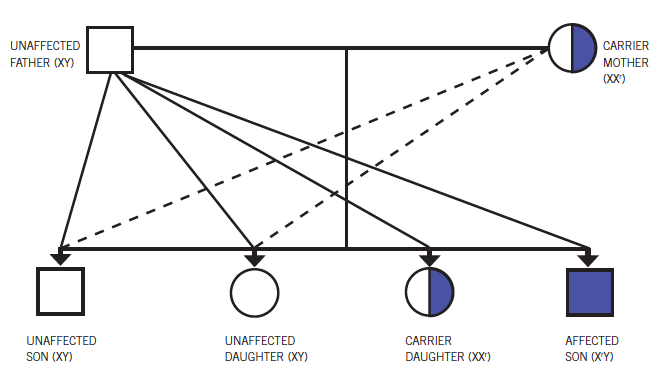
Figure 4. Becker muscular dystrophy has an X-linked recessive inheritance pattern.
Clinical Features
While DMD usually is diagnosed by the age of 3 to 5 years,1,8 the onset of weakness and hypertrophy of the same muscles in BMD occurs later, at up to 21 years of age, with a mean age at onset of 11 years.7 Dystrophin protein is expressed in skeletal, cardiac, and smooth muscle, and in the brain.
Classic features include progressive proximal muscle weakness, delayed walking, tiptoe walking, frequent falls, difficulty climbing, a waddling or Trendelenburg gait, and demonstration of the Gowers sign. Other classic features are hypertrophy or pseudohypertrophy of the calf muscles and wasting of the thigh muscles; scoliosis and/or lordosis; progressive contractures of the muscles of the ankles, knees, hips, and elbows; and connective tissue proliferation in muscle tissue.1,8
Muscle wasting begins proximally in the pelvis and lower extremities, and slowly progresses to the shoulders and neck, followed by the arm muscles and finally the respiratory muscles. Progression to the respiratory muscles can cause weak cough, frequent respiratory infections, and a decrease in respiratory reserve. Extraocular muscle function generally is well preserved, however, and sphincter incontinence occurs uncommonly and as a late event. Cardiomyopathies are frequent with dystrophinopathies, occurring in up to 80% of affected persons by second decade of life and 100% by adulthood.3 The severity of cardiac involvement does not correlate with the degree of skeletal muscle involvement; some patients with BMD may die as a result of cardiomyopathies and yet be completely ambulatory until the time of death.
Intellectual impairment is less common in BMD than in DMD. The potential for seizures is somewhat higher in persons with dystrophinopathies compared with general population.

Diagnosis
Early diagnosis is crucial to improving the survival and quality of life for persons with BMD. Dystrophinopathies should be suspected in children with no family history who are not walking by the age of 16 to 18 months; in children who demonstrate the Gowers sign at any age, but especially before the age of 5 years; in patients with unexplained elevated liver transaminase levels; and in children with a family history positive for muscular dystrophy and any suspicion of abnormal muscle function.
Assessment of serum CK levels is the standard screening test.7 CK levels usually are elevated to 15,000 to 35,000 IU/L. Normal CK levels are incompatible with the diagnosis of BMD. Electromyography results show weakness caused by destruction of nerves.7
To confirm a BMD diagnosis, dystrophin genetic testing in blood is required, even after positive muscle biopsy results. In the case of positive results of genetic testing, a muscle biopsy may be optionally performed to distinguish DMD from other phenotypes.7 Once the diagnosis is confirmed, cardiac evaluation should take place.
Management
Although there is no cure for BMD, advances in medical supportive care have increased survival time and improved quality of life for persons with it. A multidisciplinary team approach is crucial to management of the condition.7
Subspecialty consultation is necessary for regular surveillance:
• A neurologist helps establish the diagnosis and the appropriate medical management.
• A cardiologist provides surveillance and assessment of cardiomyopathies and cardiac rhythm abnormalities. Eligible symptomatic patients with BMD may eventually undergo cardiac transplantation.
• A pulmonologist provides regular pulmonary function tests, especially after a patient becomes wheelchair-bound. Weakness of the diaphragm may not parallel the involvement of skeletal or cardiac muscle. Patients with BMD therefore must be evaluated for the need for chest physical therapy, mechanical insufflation-exsufflation (cough-assist) devices, and nocturnal positive-airway pressure ventilation devices, because these interventions also may help prevent heart failure.
• A medical geneticist and/or genetic counselor offers genetic testing of the mother and siblings of a person diagnosed with BMD and counseling about the potential transmission to future children.
• An orthopedic surgeon assesses any spinal deformity and muscle contractures.
• Physical therapists, occupational therapists, and speech-language pathologists can address the functional needs of the patient as the disease progresses.7 They can help maintain functional mobility and independence in activities of daily living, provide and instruct about the use of assistive devices, and help prevent dysphagia-related complications as weakness progresses to the pharyngeal muscles.
The only clinically proven pharmacologic treatments are glucocorticoids such as prednisone, which increase muscle strength and prolong the ability to ambulate.2 However, complications secondary to corticosteroid use must be vigilantly managed. Although their use remains controversial, angiotensin-converting enzyme inhibitors and β-blockers have been proposed as a therapy to delay and treat dilated cardiomyopathy. Gene therapy, myoblast therapy, and stem cell therapy are all promising potential therapies under scientific study.2
Follow-up
Yearly influenza vaccination and all scheduled pediatric immunizations are recommended for children with BMD. Preserving a good nutrition status and avoiding obesity are important in maintaining functional mobility as muscle weakness progresses. Continuous cardiopulmonary follow-up is necessary.7
Prognosis
Complications of BMD include progressive disability; dilated cardiomyopathy and dysrhythmia; respiratory failure with the potential need for ventilator support; muscle contractures; kyphoscoliosis; dysphagia and aspiration pneumonia; functional constipation; osteopenia and pathologic fracture; and malignant hyperthermia.6-8
The prognosis is variable; some patients may be ambulatory into adult life. The mean age at which BMD patients become nonambulatory is 27 years, with a range of 12 to 30 years. Death usually results from respiratory failure or cardiac failure at a mean age of 42 years, with a range of 23 to 63 years.7
Although DMD and BMD are essentially the same phenotypically, the quality of life associated with either condition differs, a fact that is important in counseling the patient and family about living with BMD.
References:
1. Bushby K, Finkel F, Birnkrant DJ, et al; DMD Care Considerations Working Group. Diagnosis and management of Duchenne muscular dystrophy, part 1: diagnosis, and pharmacological and psychosocial management. Lancet Neurol. 2010;9(1):77-93.
2. Beytía Mde LA, Vry J, Kirschner J. Drug treatment of Duchenne muscular dystrophy: available evidence and perspectives. Acta Myol. 2012;
31(1):4-8.
3. Doo KH, Ryu HW, Kim SS, et al. A case of Becker muscular dystrophy with early manifestation of cardiomyopathy. Korean J Pediatr. 2012;
55(9):350-353.
4. Becker PE, Kiener F. A new x-chromosomal muscular dystrophy [in German]. Arch Psychiatr Nervenkr Z Gesamte Neurol Psychiatr. 1955;
193(4):427-448.
5. Becker PE. New results of genetics of muscular dystrophy [in German]. Acta Genet Stat Med. 1957;7(2):303-310.
6. The muscular dystrophies. In: Ropper AH, Samuels MA. Adams and Victor’s Principles of Neurology. 9th ed. New York, NY: McGraw-Hill; 2009:chap 50.
7. Mandac BR. Becker muscular dystrophy. Medscape Reference Web site. http://emedicine.medscape.com/article/313417-overview. Updated November 28, 2011. Accessed July 24, 2013.
8. Sarnat HB. Duchenne and Becker muscular dystrophies. In: Kliegman RM, Stanton BF, St. Geme JW III, Schor NF, Behrman RE, eds. Nelson Textbook of Pediatrics. 19th ed. Philadelphia, PA: Elsevier Saunders; 2011:2119-2122.
Dr Taylor is in the neonatal-perinatal fellowship program in the Division of Neonatology at Harbor–UCLA Medical Center in Torrance, California. Dr Peter is a clinical associate professor at the Florida State University College of Medicine and a pediatrician at Crestview Pediatrics and Adolescent Center in Crestview, Florida.