
Genetics in Gastroenterology: What You Need to Know, Part 1
ABSTRACT: Many gastrointestinal diseases are inherited or have a genetic predisposition involved in disease expression. Recent research has uncovered the genes responsible for many of these conditions. Some conditions now have genetic testing available for diagnosis and for identifying asymptomatic family members. Certain genes have been associated with other diseases, but the development of the condition is not fully understood. These developments will continue to change how we diagnose and treat gastrointestinal conditions.
Key words: gastrointestinal disease, genetic disorders, adenomatous polyposis coli syndrome, hereditary nonpolyposis colorectal cancer syndrome, juvenile polyposis, hereditary hemochromatosis, polycystic liver disease, autoimmune hepatitis, Budd-Chiari syndrome, alpha-1-antitrypsin deficiency, Wilson disease.
__________________________________________________________________________________________________________________________
The modern age of genetics began in the mid-19th century, and many scientific discoveries led to the sequencing of the human genome. Recent research has uncovered the genetic basis of many diseases, and continued study is certain to lead to more discoveries.
The results of this research will continue to change medicine. Genetic information can be used to diagnose conditions and identify asymptomatic family members so that necessary screening or treatments can be implemented. It is also important in family planning for many persons who may carry disease-causing genes. As this field continues to progress, patients will be eager to know what is available and how this can be applied to their families.
Many diseases involving the gastrointestinal organs are inherited or have a genetic predisposition involved in disease expression. Some of these have commercially available tests that can be used for diagnostic or screening purposes in predisposed individuals. Many others have a genetic component that has been identified.
These discoveries are already changing how we diagnose certain conditions and how we screen family members of patients with these conditions. In the future, we may be able to use this information to individually tailor therapies and improve outcomes and quality of life. The purpose of this 2-part article is to review this information and to discuss the implications for your practice. Here, in part 1, we address inherited colon cancer syndromes, juvenile polyposis, hereditary hemochromatosis, polycystic liver disease, autoimmune hepatitis, Budd-Chiari syndrome, alpha-1-antitrypsin deficiency, and Wilson disease. In a coming issue, we will focus on pancreatitis, celiac sprue, Peutz-Jeghers syndrome, neuroendocrine tumors, hereditary hemorrhagic telangiectasia, and inflammatory bowel disease.
INHERITED COLON CANCER SYNDROMES
The inherited colon cancer syndromes represent approximately 3% to 5% of colon cancers. They are all inherited in an autosomal dominant pattern with almost 100% penetrance.1 They are divided into adenomatous polyposis coli (APC) syndrome and hereditary nonpolyposis colorectal cancer (HNPCC) syndrome.
Adenomatous polyposis coli syndrome. APC syndrome is responsible for about 1% of the total number of colorectal cancers. The two major types of the syndrome are familial adenomatous polyposis (FAP) and Gardner’s syndrome, which are distinguished by clinical features. Both are characterized by the presence of hundreds to thousands of adenomas in the colon. Gardner’s syndrome is also characterized by the presence of extraintestinal manifestations. Patients may have osteomas, skin and soft tissue tumors, desmoid tumors, and thyroid tumors in association with multiple colon polyps.2
FAP syndrome affects nearly 1 in 8000 persons.3 The majority of patients who have FAP syndrome start to develop colonic adenomas during childhood, with 95% affected by age 35.4 Cancers start to arise about 8 to 10 years after adenomas develop; thus, most patients have colon cancer by 40 to 50 years of age. The diagnosis of FAP syndrome is based on a suggestive family history and clinical findings; genetic testing should be performed whenever possible to confirm the diagnosis. Genetic testing can also be used to screen for a presymptomatic diagnosis in individuals with a family history of FAP syndrome.
The disorder is caused by a germline mutation in the APC gene, located on chromosome 5q21.2 The APC gene is a tumor suppressor gene, and inactivation of the gene product is the initial step in the development of colorectal cancer. It was first localized in 1987 and cloned in 1991. The development of the in vitro synthesized-protein assay made genetic testing feasible, and it was introduced commercially in 1994.5 The test can detect APC gene mutations in affected individuals in about 80% of families with FAP syndrome. When the mutation in a family is known, it can differentiate affected from unaffected individuals almost 100% of the time. More than 50,000 people in the United States alone could benefit from genetic testing.3 When used properly, the test can justify frequent endoscopic surveillance for cancer risk, prophylactic colectomy when needed, and family planning for those affected.4
Once the diagnosis is made, cancer prevention is the main goal, and frequent screening with colonoscopy is started at age 16 to 18 years. Usually by the late teens or early twenties, depending on the polyp burden, prophylactic colorectal surgery is recommended to prevent cancer development.
In addition to colorectal polyps, adenomatous polyps are found in the stomach and duodenum in patients with FAP. They are especially frequent in the periampullary area of the duodenum, and they can develop into adenocarcinoma.5 After colectomy is performed, periampullary carcinoma becomes the most common malignancy, occurring in about 5% to 6% of these patients. Because of this, upper endoscopy with a gastroscope and a side-viewing duodenoscope is performed every 1 to 5 years, depending on the polyp burden.
Hereditary nonpolyposis colorectal cancer. HNPCC is a colon cancer syndrome that is inherited in an autosomal dominant fashion.6 It accounts for approximately 2% to 7% of all colorectal cancers. The syndrome is characterized by the early development of colon cancer, usually in the early to mid-40s, although it can occur in the teens or 20s. Tumors have a right-sided colon predominance, and there is a high incidence of synchronous and metasynchronous lesions. Families with this disorder are also at risk for endometrial, ovarian, gastric, small intestine, brain, urinary tract, and biliary tract tumors. These patients do not necessarily have an increased number of colon polyps, but their polyps are usually larger and have more aggressive histology.
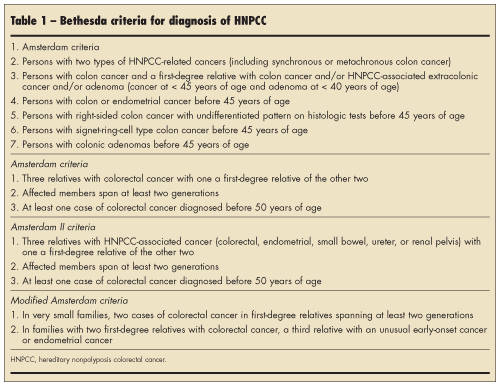
Because of the risk of early colorectal and other cancers, it is important to identify families with HNPCC syndrome. Several attempts have been made to establish clinical criteria for making the diagnosis.7 The Amsterdam criteria were established in 1991 to aid in identification of families with HNPCC and to make diagnosis more uniform. It requires families to have three affected individuals over two generations, with one being a first-degree relative of the other two and one younger than 50 years of age. There are some shortcomings, however. Sometimes multiple generations are not affected because of small family size, variable penetrance, new mutations, or incomplete family information. Amsterdam II criteria consider extracolonic manifestations. The Bethesda criteria (Table 1) have since been established and have a more liberal definition of HNPCC. Identification of these families is necessary so that surveillance for colorectal cancer can be performed.
HNPCC is caused by germline mutations in DNA mismatch repair (MMR) genes.7 These MMR genes identify and correct mismatched base pairs of DNA.8 Over 90% of HNPCC cases are caused by germline mutations on hMSH2 or hMLH1, but there is no uniform type of mutation. The abnormalities can lead to a broad spectrum of truncating, frameshift, and missense mutations.
Commercial genetic testing for both hMSH2 and hMLH1 was introduced in the 1990s. The test is performed on DNA isolated from peripheral blood mononuclear cells and, because full gene sequencing is required, is expensive. The recommendation is to first test a patient who meets the clinical criteria of HNPCC. If the genetic results are consistent with the disease, at-risk family members can then be tested for the presence of the mutation found in the index case. However, some families meet the clinical criteria for HNPCC but have normal genetic test results, which does not rule out the disease.
Patients with HNPCC syndrome have an 80% to 85% lifetime risk of developing colorectal cancer.7 Screening colonoscopy is recommended every 2 years starting at age 25 and annually after age 40. Detecting this condition is important so that patients can take preventive measures.
JUVENILE POLYPOSIS
Juvenile polyposis is an autosomal dominant condition found in one in 100,000 persons.9 Patients usually develop juvenile polyps in the gastrointestinal tract by the third decade of life, which primarily affect the colon and rectum.10 Patients with juvenile polyposis are at increased risk for colorectal cancer. Gastric, small bowel, and pancreatic cancers have been reported. Juvenile polyposis has been linked to chromosome 18q21 with MADH4 germline mutations.9 Other cases have been linked to chromosome 10q22-23 with a germline mutation in the bone morphogenetic protein receptor 1A (BMPR1A) gene. Because these mutations do not explain all cases of juvenile polyposis, there may be other genes responsible for the disorder.
 HEREDITARY HEMOCHROMATOSIS
HEREDITARY HEMOCHROMATOSIS
Hereditary hemochromatosis is the most common genetic disorder in the Caucasian population.11 Its geographic distribution is worldwide, but it is most common in persons of northern European origin. The prevalence is close to 1 per 200 in those with Nordic or Celtic ancestry.
Hemochromatosis affects the liver, heart, pancreas, and endocrine system.12 Presenting symptoms include fatigue, skin darkening (Figure 1), hair loss especially in the axillae, impotence, and arthralgias. Heart failure, arrhythmias, and diabetes can also occur and are associated with a worse prognosis. The disease may present as liver fibrosis or cirrhosis and hepatocellular carcinoma, which accounts for about one third of deaths in affected patients. Hypogonadism and adrenocortical failure can also occur as a result of iron overload, but they usually occur in young patients with pathologic iron storage.
When iron overload is suspected, a complete history is taken to include blood transfusions, use of iron or other supplements, family history, ethnic origin, diet, and alcohol use.12 Measurements of serum ferritin, transferrin, iron, and transferrin saturation or total iron-binding capacity should be performed. Elevated transferrin saturation predicts the presence of hemochromatosis.11 Transferrin saturation is derived by dividing fasting serum iron by total iron-binding capacity and multiplying by 100. A value of greater than 50% in women and greater than 60% in men has a sensitivity of 0.92, a specificity of 0.93, and a positive predictive value of 86% for the diagnosis of hereditary hemochromatosis. Performing these tests fasting eliminates 80% of false positive results. If a screened individual has an elevated transferrin saturation and serum ferritin, genetic testing is indicated.
The most common form of hereditary hemochromatosis is an autosomal recessive inherited mutation in the HFE gene which is located in the MHC region on chromosome 6.13 It is a single base change that results in the substitution of tyrosine for cysteine at position 282 of the HFE protein (282Y). It seems to have originated in a single Celtic or Viking ancestor in northwestern Europe about 2000 years ago. It caused no obstacle to reproduction and was spread by population migration. The defect may have had some advantages, such as resistance to dietary iron deficiency and certain infectious diseases. C282Y mutation homozygosity is now found in about 5 in every 1000 persons of northern European descent, which is 10 times more common than cystic fibrosis. The inheritance follows classic Mendelian genetics. About 90% to 95% of HFE-related hemochromatosis is from C282Y homozygosity. C282Y/H63D compound heterozygotes contribute 4%. Those with H63D homozygosity may have elevated transferrin saturation or serum ferritin levels but do not develop serious iron overload.
Phenotypic expression in hereditary hemochromatosis is variable and incomplete. About 75% of C282Y homozygotes have biochemical expression with elevated serum ferritin levels, but the proportion that develops clinically significant disease is highly controversial. Environmental factors and genetic modifiers likely interact to influence penetrance and expression of genotype.
The HFE mutations C282Y and H63D can be detected by commercially available polymerase chain reaction assays. First-degree relatives of identified C282Y homozygotes should also be tested. Because organ damage does not occur until adulthood, evaluation of relatives can be postponed until they are 20 years of age.
In a small proportion of patients, iron overload may be caused by ferroportin or transferrin receptor 2 mutations. Juvenile hemochromatosis is caused by genetic mutations in hemojuvelin and hepcidin. Rarely, these mutations can coexist in patients with C282Y mutations. These patients appear to exhibit more severe iron overload than those without hemojuvelin and hepcidin mutations. These mutations are rare and account for only a minor portion of variability of expression of the HFE genotype. Commercial assays to detect these mutations are not currently available. Therefore, it is likely that significant unidentified genetic factors have yet to be elucidated.
Liver biopsy is used to document the presence of cirrhosis and to rule out significant iron overload when iron markers are equivocal.11 It can also be used to investigate other possible causes of liver disease. Extensive bridging fibrosis or cirrhosis found on liver biopsy has a profound impact on prognosis. Liver biopsy should be offered to all C282Y homozygotes over the age of 40 or those with elevated serum alanine transaminase levels, clinical evidence of liver disease, or serum ferritin greater than 1000 ng/mL.
If patients receive an early diagnosis and are treated, survival in those without cirrhosis is similar to the normal population. In contrast, mortality is significantly increased in those with cirrhosis even after they undergo successful phlebotomy. Cirrhosis and its complications, particularly hepatocellular cancer, account for three quarters of deaths related to hereditary hemochromatosis.
The mainstay of treatment of hemochromatosis is phlebotomy.11 Early identification and preemptive treatment of those at risk is required. Institution of treatment before cirrhosis and diabetes develop significantly reduces morbidity and mortality. Asymptomatic patients with homozygous hereditary hemochromatosis and markers of iron overload, others with potentially toxic levels of hepatic iron, and symptomatic patients should all be treated. Treatment of symptomatic patients is advocated to mitigate as much of the organ damage as possible. Malaise, fatigue, skin pigmentation, insulin requirement in diabetes, and abdominal pain may be reduced by phlebotomy. Arthropathy, hypogonadism, and cirrhosis are less responsive to therapy. Complications of cirrhosis, particularly hepatocellular carcinoma, continue to increase mortality, even with treatment, but can be prevented if the condition is diagnosed early and phlebotomy started before the development of cirrhosis.
POLYCYSTIC LIVER DISEASE
Polycystic liver disease encompasses disorders associated with cysts only in the liver and those associated with cysts in the liver and kidneys.14 Autosomal dominant polycystic kidney disease (ADPKD) is associated with cysts in both organs and is associated with PKD1 and PKD2. Autosomal dominant polycystic liver disease (ADPLD) is characterized by liver cysts only and is associated with protein kinase C substrate 80K-H and SEC63.
ADPKD is the most common inherited nephropathy and affects 0.2% of the population. Affected persons experience progressive development and enlargement of fluid-filled cysts in the kidneys, causing renal failure in 50%. The liver becomes polycystic at a late stage. ADPKD is caused by a loss-of-function mutation in either PKD1 or PKD2. The PKD1 gene is found on the short arm of chromosome 16p13.3-p13.12 and encodes the protein polycystin-1.15 This protein is a plasma-membrane receptor involved in regulating several biological functions and signaling pathways.14 The PKD2 gene is found on chromosome 4q21-q23 and encodes the protein polycystin-2.15 This protein functions as a Ca2+-permeable cation channel and belongs to the transient receptor potential family of ion channels.14
ADPLD occurs in approximately 1 in 100,000 persons; those who are affected have few or no renal cysts.14 The two genes that have been located are responsible for one-third to one-half of the ADPLD cases. PRKCSH is found on chromosome 19p13.2 and encodes the protein kinase C substrate 80K-H or hepatocystin. Hepatocystin is located in the endoplasmic reticulum in a variety of organs and is involved in the maturation and folding of glycoproteins. Mutations result in the absence of the C-terminal part of hepatocystin, which keeps it from attaching to the endoplasmic reticulum. SEC63 is located on chromosome 6q21 and encodes the Sec63 protein. The Sec63 protein is part of the protein-translocation machinery in the endoplasmic reticulum and is involved in processing newly made glycoproteins. The way in which these genetic mutations lead to cyst formation has not been elucidated.
There is no medical therapy for liver cysts.15 Cyst aspiration and sclerosis can be performed if there are one or a few dominant cysts, but recurrence is common. Cyst fenestration and partial liver resection are surgical approaches that can be performed for massive cystic disease. Patients with cysts that are refractory to other treatments can be considered for liver or combined liver-kidney transplant.
AUTOIMMUNE HEPATITIS
Autoimmune hepatitis is an inflammatory condition of the liver characterized by interface hepatitis, portal plasma cell infiltration, hypergammaglobulinemia, and autoantibodies.16 Women are more commonly affected and all ethnic groups are susceptible, although those of Northern European ancestry are at highest risk. It is caused by a complex interaction between triggering factors, autoantigens, genetic predispositions, and immunoregulatory networks. Clinical manifestations are variable. Patients may present with nonspecific symptoms such as fatigue, malaise, anorexia, abdominal pain, itching, and arthralgias.17 Some may present with fulminant disease, manifested by profound jaundice, coagulopathy, and aminotransferase levels in the thousands. While a diagnosis is commonly established based on the presence of autoantibodies and typical liver biopsy findings, genetic changes may modulate the severity of the disease.
The HLA genes in the major histocompatibility complex on the short arm of chromosome 6 appear to play the dominant role in predisposing to autoimmune hepatitis.17 Type 1 autoimmune hepatitis is associated with the HLA-DR3 and HLA-DR4 serotypes. Patients with this type have circulating ANA, smooth muscle antibodies, antiactin antibodies, atypical pANCA, and autoantibodies against soluble liver antigen and liver-pancreas antigen. HLA-DR3 is associated with more severe disease, usually in young women. Type 2 autoimmune hepatitis is associated with the HLA-DRB1 and HLA-DQB1 alleles. Type 2 is rare and is characterized by antibodies against liver-kidney microsome 1 and liver cytosol 1.
An association with tumor necrosis factor genes found in the class III region of the MHC has been suggested, but the exact relationship is unclear.17 Other possible genetic factors are under investigation.
BUDD-CHIARI SYNDROME
The Budd-Chiari syndrome is a heterogeneous group of disorders characterized by hepatic venous outflow obstruction.18 It can occur at the level of the hepatic venules, large hepatic veins, inferior vena cava, or right atrium. Some patients are asymptomatic, but others can present with fulminant, acute, subacute, or chronic disease.
Hereditary and acquired factors causing a hypercoagulable state predispose to the development of Budd-Chiari syndrome, and they can be found in about 75% of cases. Myeloproliferative disorders are the most common cause. There is a prevalence of the Janus kinase 2 tyrosine kinase mutation V617F in patients with myeloproliferative disorder and Budd-Chiari syndrome.19 JAK2 intermediates between growth factor receptors on the hematopoietic progenitor cell surface and cytoplasmic signaling molecules. It has an active kinase domain (Jak homology 1) that is negatively regulated by an active pseudokinase domain (Jak homology 2). A guanine-to-thymine mutation encoding a valine-to-phenylalanine substitution at position 617 in the JH2 domain affects the autoinhibitory function.
 ALPHA-1-ANTITRYPSIN DEFICIENCY
ALPHA-1-ANTITRYPSIN DEFICIENCY
Alpha-1-antitrypsin (AT) deficiency is found in 1:1800 live births.20 It is usually first recognized in an infant who presents with prolonged obstructive jaundice. Patient may then develop pulmonary and/or liver manifestations, but phenotypic expression is variable.21 Chronic obstructive pulmonary disease (COPD), especially emphysema, is the most common clinical manifestation of AT deficiency, and it is worsened by smoking. Patients usually develop COPD between the ages of 40 and 50 years if they smoke. Liver manifestations, besides jaundice, usually present in adulthood and include cirrhosis and fibrosis. Hepatocellular carcinoma can occur in these patients. AT deficiency can also cause vascular disease such as intracranial aneurysms, panniculitis, anterior uveitis, systemic necrotizing vasculitis, and Wegener granulomatosis.
AT deficiency can be detected by demonstration of a low plasma level of alpha-1-antitrypsin and evaluation of the protein by protease inhibitor (PI) typing. The PI*M allele is the most common in all populations. The PI*Z allele is the most common allele that causes deficiency and the PI*S is another allele that can cause deficiency and is more prevalent in certain populations. An individual who is PI MM is normal and has a normal plasma concentration of alpha-1-antitrypsin. A patient with PI MZ has a slightly increased risk of lung disease. PI SZ is not usually associated with liver or lung disease alone, but smokers are at higher risk for COPD. Those with PI ZZ have very low plasma levels of alpha-1-antitrypsin and are at risk for clinical disease.
AT deficiency is an autosomal recessive disorder in which a point mutation causes the ATZ molecule to have an abnormal folding pathway and aggregation.20 Instead of being secreted into the blood and body fluid to inhibit neutrophil elastase activity, it is retained in the endoplasmic reticulum of hepatocytes. The gene responsible is serpin peptidase inhibitor, clade A, member 1, SERPINA1, which encodes alpha-1-antitrypsin, and homozygotes will have mutations in both copies of the gene. The test is available for confirmatory diagnosis, carrier testing, and prenatal diagnosis.
WILSON DISEASE
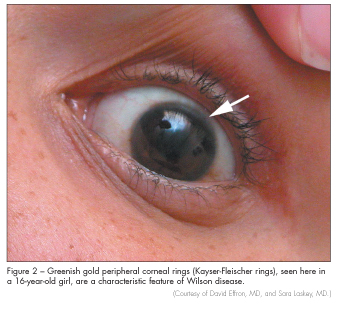
Wilson disease is found in 1 in 30,000 persons and is characterized by hepatic and neurologic manifestations of copper accumulation.22 Patients can present with hepatitis, cirrhosis, or liver failure.23 Neurologic manifestations include movement disorders, slurred speech, tremor, dystonia, and Parkinson-like symptoms. Patients often have behavioral abnormalities before other clinical manifestations. Wilson disease should be suspected in young patients with these symptoms and signs. If it is suspected, serum ceruloplasmin level and 24-hour urine copper excretion should be measured. Kayser-Fleischer rings (Figure 2) on slit lamp examination may also be seen.
Wilson disease is autosomal recessive.23 The responsible gene is ATP7B, which codes for a membrane-bound, copper-binding protein that is expressed primarily in the liver and there are many causative mutations. The liver is responsible for copper balance regulation, and excess copper is excreted in the bile. Mutations in the ATP7B gene disrupt this pathway. Because multiple mutations lead to Wilson disease, genetic testing is not commonly used to diagnose the disease. Once a mutation is established in an affected patient, testing of first-degree relatives is feasible.

GENETIC TESTING AND COUNSELING
Medicine has entered a new era, with many genetic tests available to assist clinicians. Many tests are already commercially available (Table 2), but many more are certain to follow. Physicians can now obtain detailed genetic information on patients, which is leading to personalized medicine. This information can be used for prenatal diagnosis, newborn screening, carrier screening, diagnosis, and pharmacogenetics.24 As information about these tests becomes increasingly available, patients will surely have many questions regarding these tests and how they can be of benefit.
Genetic counseling should be offered with genetic testing. There are psychological effects in knowing that one is genetically predisposed to certain cancers and other life-altering conditions. This may lead to depression and changes in quality of life and can cause patients to avoid having children. This may also affect patients’ ability to obtain health or life insurance, although they should be legally protected. There are times when this information may be more harmful than helpful; thus, the decision to perform genetic testing should be handled on an individual basis.
1. Ahnen DJ. Genetics of colon cancer. West J Med. 1991;154:700-705.
2. Galiatsatos P, Foulkes WD. Familial adenomatous polyposis. Am J Gastroenterol. 2006;101:385-395.
3. Powell SM, Petersen GM, Krush AJ, et al. Molecular diagnosis of familial adenomatous poly-posis. N Engl J Med. 1993;329:1982-1987.
4. Half E, Bercovich D, Rozen P. Familial adenomatous polyposis. Orphanet J Rare Diseases. 2009;4:22.
5. Giardiello FM, Brensinger JD, Petersen GM, et al. The use and interpretation of commercial APC gene testing for familial adenomatous polyposis.
N Engl J Med. 1997;336:823-827.
6. Halbert CH, Lynch H, Lynch J, et al. Colon cancer screening practices following genetic testing for hereditary nonpolyposis colon cancer mutations. Arch Intern Med. 2004;164:1881-1887.
7. Robinson KL, Liu T, Vandrocova J, et al. Lynch syndrome (hereditary nonpolyposis colon cancer) diagnostics. J Natl Cancer Inst. 2007;99:291-299.
8. Ghung DC, Rustgi AK. The hereditary nonpoly-posis colorectal cancer syndrome: genetics and clinical implication. Ann Intern Med. 2003;138:560-570.
9. Howe JR, Sayed MG, Ahmed AF, et al. The prevalence of MADH4 and BMPR1A mutations in juvenile polyposis and absence of BMPR2, BMPR1B, and ACVR1 mutations. J Med Genet. 2004;41:484-491.
10. Brosens LA, van Hattem A, Hylind LM, et al. Risk of colorectal cancer in juvenile polyposis. Gut. 2007;56:965-967.
11. Tavill AS. Diagnosis and management of hemochromatosis. Hepatology. 2001;33(5):1321-1328.
12. Zoller H, Cox TM. Hemochromatosis: genetic testing and clinical practice. Clin Gastroenterol Hepatology. 2005;3:945-958.
13. Pietrangelo A. Hereditary hemochromatosis: pathogenesis, diagnosis, and treatment. Gastroenterology. 2010;139:393-408.
14. Temmerman F, Missiaen L, Bammens B, et al. Systematic review: the pathophysiology and management of polycystic liver disease. Aliment Pharmacol Ther.2011;34:702-713.
15. Everson GT, Taylor MRG, Doctor RB. Polycystic disease of the liver. Hepatology. 2004;40:774-782.
16. Czaja AJ, Freese DK. Diagnosis and treatment of autoimmune hepatitis. Hepatology. 2002;36(2):479-497.
17. Krawitt EL. Autoimmune hepatitis. N Engl J Med. 2006;354:54-66.
18. Menon KV, Shah V, Kamath PS. The Budd-Chiari syndrome. N Engl J Med. 2004;350:578-585.
19. Patel RK, Lea NC, Heneghan MA, et al. Prevalence of the activating JAK2 tyrosine kinase mutation V617F in the Budd-Chiari syndrome. Gastroenterology.2006;130:2031-2038.
20. Perlmutter DH, Brodsky JL, Balistreri WF. Trapnell BC. Molecular pathogenesis of alpha-1-antitrypsin deficiency-associated liver disease: a meeting review.Hepatology. 2007;45:1313-1323.
21. Schlade-Bartusiak K, Cox DW. Alpha-1-antitrypsin deficiency. NCBI Bookshelf. 2008.
22. Gow PJ, Smallwood RA, Angus PW, et al. Diagnosis of Wilson’s disease: an experience over three decades. Gut. 2000;46:415-419.
23. Brewer GJ. Recognition, diagnosis, and management of Wilson’s disease. PSEBM. 2000;223:39-46.
24. Lerman C, Croyle RT, Tercyak KP, Hamman H. Genetic testing: psychological aspects and implications. J Consulting Clin Psychology. 2002;70(3):784-797.