
Peer Reviewed
Frequency and Clinical Characteristics of Abnormal Sucrase Enzyme Activity in Pediatric Patients
AUTHORS:
Amanda Tran, BA1 • Vrinda Bhardwaj, MD1,2 • Tanaz Danialifar, MD1,2
AFFILIATIONS:
1Keck School of Medicine, University of Southern California, Los Angeles, California
2Department of Pediatric Gastroenterology, Hepatology, and Nutrition, Children’s Hospital Los Angeles, Los Angeles, California
CITATION:
Tran A, Bhardwaj V, Danialifar T. Frequency and clinical characteristics of abnormal sucrase enzyme activity in pediatric patients. Consultant. 2022;62(7):e1-e5. doi:10.25270/con.2021.11.00012
Received July 18, 2021. Accepted August 4, 2021. Published online November 19, 2021.
DISCLOSURES:
Bhardwaj and Tran declare no competing interests. Danialifar is a consultant and speaker for QOL Medical, LLC, and has received nongrant funding support from QOL Medical.
CORRESPONDENCE:
Amanda Tran, BA, Keck School of Medicine, 1975 Zonal Avenue, Los Angeles, CA 90033 (amandalt@usc.edu)
ABSTRACT
Objective. To determine the frequency and clinical characteristics of abnormal sucrase activity in pediatric patients undergoing esophagogastroduodenoscopy (EGD) and disaccharidase analysis.
Methods. A retrospective chart review was performed for pediatric patients who underwent EGD and duodenal biopsy with disaccharidase analysis between May 2017 to May 2018 at Children’s Hospital, Los Angeles. Statistical analyses examined the associations between disaccharide activity and clinical characteristics, including gastrointestinal symptoms, existing primary gastrointestinal disease, and medication use prior to biopsy.
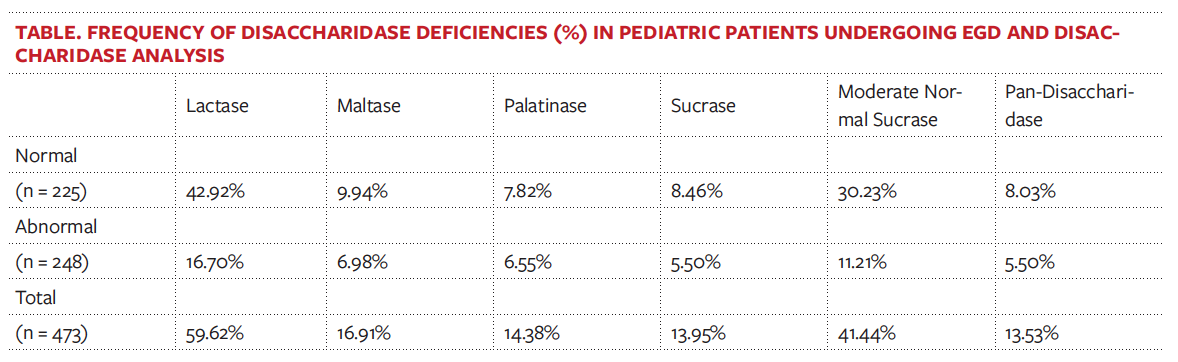
Results. A total of 473 biopsies from patients aged 4 months to 21 years were analyzed. The frequency of sucrase deficiency (< 25 μg/mL/min) was 13.95%. The frequency of moderate normal sucrase activity (25-50 μg/mL/min) was 41.44%. Of all the biopsies, 60.61% had demonstrated normal duodenal histology, and 39.39% had shown abnormal histology. Of moderate normal activity biopsies, 72.96% had shown normal histology and 27.04% had demonstrated abnormal histology. Abdominal pain (62.8%), diarrhea (28.1%), and constipation (22.5%) were commonly reported among patients with moderate normal activity.
Conclusions. Sucrase deficiency is more prevalent than previously understood and may be an underrecognized etiology of gastrointestinal symptoms in pediatric patients. A high frequency of moderate normal sucrase activity was identified, but the clinical significance remains unclear. No significant differences in symptoms were identified between patients with or without sucrase deficiency undergoing EGD. A high frequency of abdominal pain, diarrhea, and constipation was identified in patients undergoing EGD and disaccharidase analysis. Diarrhea was significantly associated with abnormal sucrase activity.
BACKGROUND
Congenital sucrase-isomaltase deficiency (CSID) caused by deficient or altered function of the sucrase-isomaltase enzyme results in accumulation of undigested sugars, carbohydrate malabsorption, osmotic diarrhea, and a host of other gastrointestinal symptoms.1 CSID was first clinically described in 1960 and has since been classically characterized by the presentation of watery diarrhea, irritability, and failure to thrive in infants between ages 9 and 18 months until the last 2 decades.2,3 Recent studies have identified sucrase deficiency in patients with an increasingly diverse array of symptoms, including recurrent abdominal pain, bloating, flatulence, diarrhea, and vomiting, suggesting that sucrase-isomaltase deficiency may be a more common and frequently misdiagnosed cause of persistent gastrointestinal symptoms.1,2,4 The frequency of sucrase-isomaltase deficiency has more recently been identified in 7.6% to 14.3% of pediatric patients undergoing esophagogastroduodenoscopy (EGD) and disaccharidase analysis.3,5-7
Primary and secondary causes of sucrase-isomaltase deficiency may produce similar clinical manifestations, and histologic evaluation of intestinal biopsy is necessary to distinguish between primary and secondary etiologies. The gold standard for diagnosis of sucrose-isomaltase deficiency is duodenal biopsy and ex-vivo analysis of disaccharidase activity.1 Using the Dahlqvist method, duodenal specimen are incubated with disaccharide substrate, and the relative disaccharidase enzyme activity is quantified as a measure of glucose produced over time.8 Sucrase deficiency is clinically defined as enzyme activity less than 25 μg/mL/min, and normal activity defined as 54.4 ± 25.4 μg/mL/min protein.9
While CSID was originally believed to be a rare, autosomal-recessive disease occurring in select populations, recent genetic and clinical studies have provided increasing evidence for variable patterns of inheritance that manifest on a clinical spectrum with varying degrees of severity.10,11 Novel compound heterozygote mutations have been identified in the sucrase-isomaltase gene that affect only a single allele and cause insufficient sucrase-isomaltase protein expression in the intestinal brush border membrane, typically causing less severe clinical manifestations than homozygous etiologies.11,12 Other studies have also found evidence that patients with common CSID mutations coding for aberrant or defective enzymes predispose patients to functional gastrointestinal disorders (FGID) such as irritable bowel syndrome and that these patients have experienced reduced symptoms from a starch- and sucrose-reduced diet.13,14 In a 2021 study, 13 different pathogenic sucrase-isomaltase gene variants were found in 29.0% of patients with abnormal sucrase activity and FGIDs, as well as in 6.4% of patients with moderate normal sucrase activity (25-55 μg/mL/min).15 Whereas previous studies have only characterized the frequency and symptoms of patients with historically defined sucrase-isomaltase deficiency, we also sought to characterize the frequency and characteristics of patients with moderate normal levels of activity (enzyme activity, 25-50 μg/mL/min).
METHODS
A retrospective chart review was performed on records of pediatric patients who had undergone EGD and duodenal biopsy with disaccharidase analysis between May 2017 to May 2018 at Children’s Hospital, Los Angeles. This study was conducted as approved by the Institutional Review Board at Children’s Hospital, Los Angeles (CHLA 1800267).
All patients who received EGD and disaccharidase analysis within the study period were included in our evaluation. Demographic data including age and race/ethnicity were included in our retrospective chart review. Clinical characteristics—which included gastrointestinal symptoms, existing diagnosis of primary gastrointestinal disease (eg, inflammatory bowel disease, short bowel syndrome, Celiac disease, eosinophilic gastrointestinal disease, irritable bowel syndrome, Helicobacter pylori infection, small intestinal bacterial overgrowth, and other), and medication use (eg, proton pump inhibitors, H2-antagonists, and probiotics) 3 months prior to biopsy—were analyzed for patients who had undergone EGD with disaccharidase analysis. Disaccharidase analysis was completed by Joli Diagnostic per the Dahlqvist Method.8
Sucrase activity was interpreted using the historical reference range for normal sucrase activity (54.4 ± 25.4 μg/mL/min protein) and sucrase deficiency (< 25.0 μg/mL/min protein).16 Moderate normal sucrase activity was defined as 25.0 to 50.0 μg/mL/min protein.
Descriptive statistics and Chi-Square tests of association were used to examine the association between disaccharidase activity and clinical characteristics. ANOVA was used to assess differences in enzyme activity across racial/ethnic groups; patients who did not specify ethnicity were excluded from this analysis.
RESULTS
A total of 466 patients aged 4 months to 21 years were included in the analysis. Of those, 7 patients had undergone evaluation twice within the specified time period, yielding a total of 473 biopsies.
The frequency of sucrase deficiency was 13.95%, and the frequency of moderate normal sucrase activity was 41.44% (Figure 1). Lactase deficiency (< 15.40 μg/mL/min) occurred in 59.62% of patients, maltase deficiency (< 100 μg/mL/min) occurred in 16.91%, palatinase deficiency (< 5 μg/mL/min) occurred in 14.38%, and pan-disaccharidase deficiency occurred in 13.53% (Table).
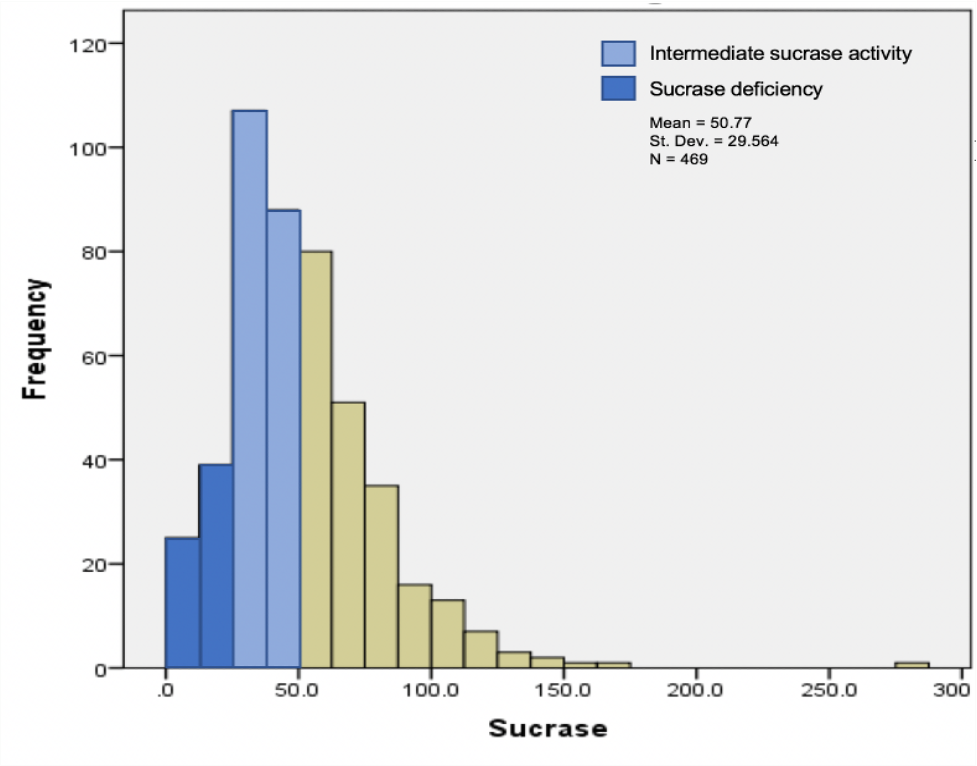
Figure 1. Frequency of pediatric patients with sucrase deficiency, moderate normal sucrase activity, and normal sucrase activity. Sucrase deficiency was defined as less than 25 μg/mL/min, moderate normal sucrase as between 25 and 50 μg/mL/min, and normal sucrase activity as more than 50 μg/mL/min.
There were no cases of isolated sucrase deficiency; all patients with sucrase deficiency had concomitant lactase deficiency. Of the patients with moderate normal sucrase, 75.5% and 7.14% had concomitant lactase and maltase deficiency, respectively. The frequency of isolated moderate normal sucrase activity was 25%.
Of the 466 patient records reviewed, race/ethnicity was specified for 318 patients. Of those, 48% were Hispanic/Latinx, 35% were White, 10% were Asian/Pacific Islander, 6% were Black, and 1% were Native American. There was no significant association between race/ethnicity and sucrase deficiency or moderate normal sucrase activity. Overall, 51.1% of patients in our study were female, and 48.9% were male. The median age of patients with moderate normal sucrase activity and sucrase deficiency was 13 years and 12 years, respectively (Figure 2).
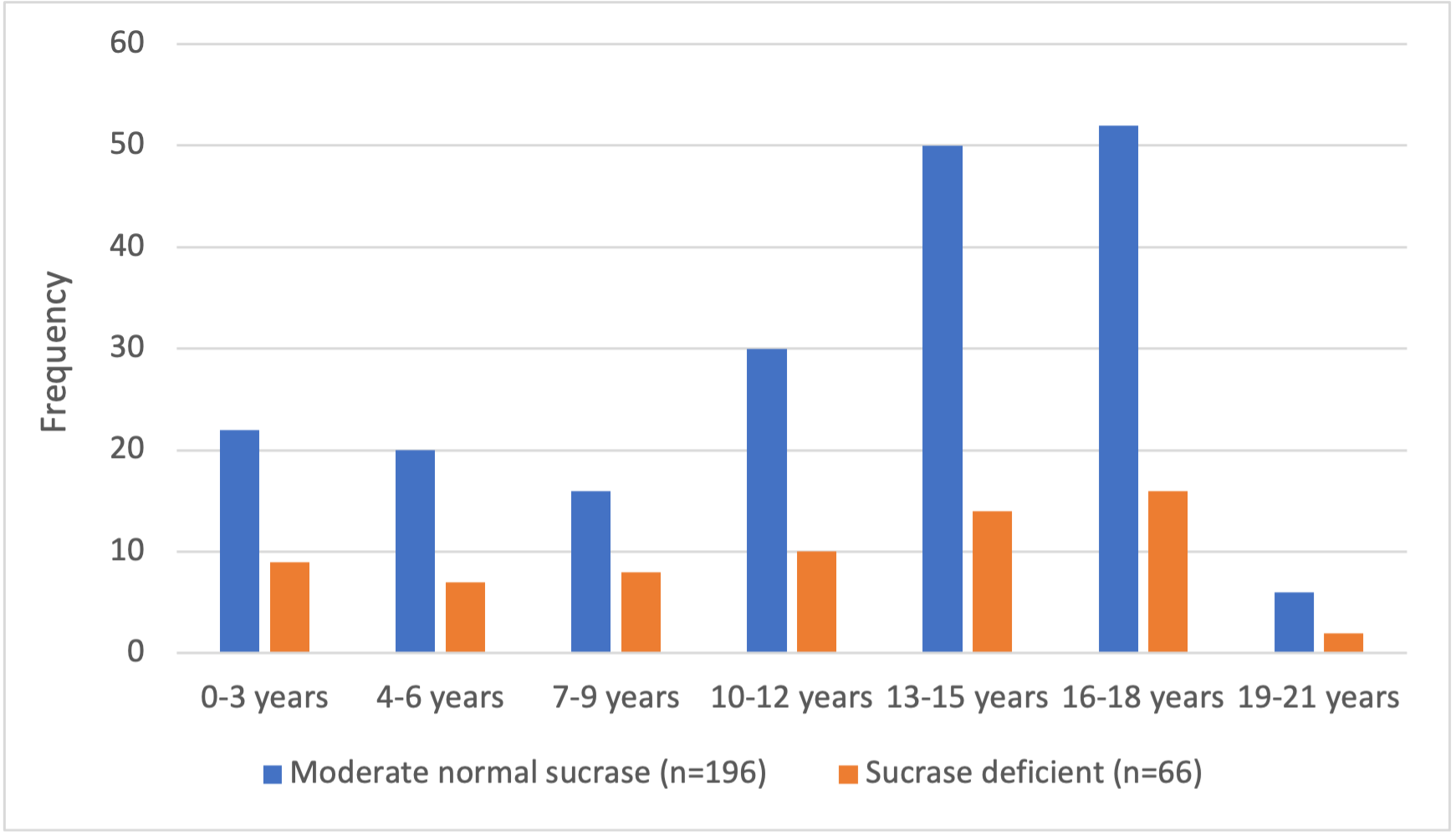
Figure 2. Age distribution of patients with moderate normal sucrase activity (n = 196) and sucrase deficiency (n = 66). The median age of patients was 13 years for moderate normal sucrase activity and 12 years for sucrase deficiency.
A total of 60.61% of biopsies with sucrase deficiency demonstrated normal duodenal histology and 39.39% demonstrated abnormal histology. Of the biopsies with moderate normal sucrase activity, 72.96% demonstrated normal duodenal histology and 27.04% demonstrated abnormal histology.
Patients in the normal, moderate, and high sucrase activity subgroups commonly presented with abdominal pain (62.4%, 62.8%, 61.9%), diarrhea (24.8%, 28.1%, 28.3%), and constipation (24.3%, 22.5%, 23.0%). No differences were identified between different sucrase activity subgroups. Diarrhea was significantly associated with abnormal sucrase activity level (p = 0.032), meaning that patients with diarrhea were more likely to have abnormal sucrase levels than patients without diarrhea. The proportion of patients with dysphagia and normal sucrase activity was significantly higher than in patients with moderate normal sucrase activity (p = 0.043).
Celiac disease was significantly associated with sucrase deficiency (p = 0.000). There was no significant association between Celiac disease and moderate normal sucrase activity. No other primary diseases were associated with sucrase deficiency or moderate normal activity.
Regarding medication use, there was a trend toward increased rates of moderate normal sucrase activity among patients taking proton pump inhibitors (p = 0.026). There was no association of proton pump inhibitor use with sucrase deficiency and no association of abnormal sucrase activity with other medications.
DISCUSSION
Previous retrospective studies that assessed sucrase deficiency in children undergoing EGD have reported a frequency of 7.6% to 14.3%.3,5-7 In our study, we found the frequency of sucrase deficiency to be 14.0%—the upper limit of the range described in the literature. The higher frequency of sucrase deficiency that we found may in part be explained by differences in the ethnic makeup of the patient population in our study compared with those of previous sucrase deficiency studies. To date, sucrase deficiency research has largely examined White, African, and Asian subjects.6 In our study, 48% of patients who specified their race/ethnicity were Hispanic/Latinx. We did not find a significant association between race/ethnicity and sucrase deficiency, but further studies are needed to characterize the relationship between race/ethnicity and sucrase deficiency, since 31% (148 of 466) of patients did not specify race/ethnicity in our study.
Unlike prior studies that have described the frequency and clinical characteristics of sucrase deficiency, we also characterized the frequency and clinical characteristics of moderate normal sucrase activity.15 Until recently, genetic studies on sucrase activity have been limited to describing sucrase-isomaltase gene variants in traditionally defined cases of sucrase deficiency. In a 2021 genetic study, pathogenic sucrase-isomaltase gene variants were identified in cases of sucrase deficiency, as well as in cases of moderate normal sucrase activity.15 Of the patients with moderate sucrase activity in this study, compound heterozygous mutations were present in 2 patients, and a few low-frequency sucrase-isomaltase gene variants were identified in normal and moderate sucrase groups that were not present in the sucrase-deficient group.15 Our findings show that up to 41.0% of pediatric patients undergoing EGD and disaccharidase analysis had moderate normal sucrase activity. Most of the moderate normal activity was found in patients with normal duodenal histology and, thus, did not attribute to secondary causes. Moderate normal sucrase activity was also associated with similar frequency of gastrointestinal symptoms to that seen in patients with sucrase deficiency, including abdominal pain, diarrhea, constipation.
Our study suggests that patients with sucrase activity between 25 and 50 μg/mL/min may comprise a subgroup of patients with defective or altered sucrase activity who may have been subjected to delayed diagnosis or delayed treatment. Additional genetic studies are needed to examine the potential relationship between the heterozygous genotype in the sucrase-isomaltase gene and a clinical phenotype marked by moderate normal sucrase function. Further research is also necessary to assess whether patients with moderate normal sucrase activity would benefit from treatment typically prescribed to patients with sucrase deficiency, including a sucrose-reduced diet and/or sacrosidase enzyme replacement therapy.
There are inherent limitations to our study given its retrospective design. There was no standardization for recording gastrointestinal symptoms, as this was collected from existing medical records. Additionally, use of disaccharidase testing was at the discretion of the treating physician. Indication for EGD and disaccharidase analysis was unclear for a few patients who had no documented gastrointestinal symptoms prior to undergoing EGD. At our institution, tissue samples are placed on ice immediately. However, it is unclear how specimen handling may have affected enzyme degradation or contributed to false-positive rates of enzyme deficiencies.
CONCLUSIONS
Our study supports that abnormal sucrase activity is more prevalent than previously thought and may be an underrecognized etiology of common gastrointestinal symptoms in pediatric patients. A high frequency of characteristic irritable bowel syndrome symptoms including abdominal pain, diarrhea, and constipation were commonly reported in pediatric patients undergoing EGD and disaccharidase analysis. A high frequency of moderate normal sucrase activity was identified, but the clinical significance remains unclear. No significant differences in symptoms were identified in patients with or without sucrase deficiency undergoing EGD. Patients undergoing EGD and disaccharidase analysis with sucrase activity greater than the historic cutoff for sucrase deficiency (< 25 μg/mL/min ) and characteristic irritable bowel syndrome symptoms may represent a subgroup of patients who may benefit from treatments typically prescribed to patients with traditional sucrase deficiency.
References
1. Gericke B, Amiri M, Naim HY. The multiple roles of sucrase-isomaltase in the intestinal physiology. Mol Cell Pediatr. 2016;3(1):2. https://doi.org/10.1186/s40348-016-0033-y
2. Treem WR. Clinical aspects and treatment of congenital sucrase-isomaltase deficiency. J Pediatr Gastroenterol Nutr. 2012;55 Suppl 2:S7-S13. https://doi.org/10.1097/01.mpg.0000421401.57633.90
3. Cohen SA. The clinical consequences of sucrase-isomaltase deficiency. Mol Cell Pediatr. 2016;3(1):5. https://doi.org/10.1186/s40348-015-0028-0
4. Robayo-Torres CC, Baker SS, Chumpitazi BP, Lecea CE, Nichols BL Jr, Opekun AR. Poor starch digestion in children with CSID and recurrent abdominal pain. J Pediatr Gastroenterol Nutr. 2012;55 Suppl 2:S32-S34. https://doi.org/10.1097/01.mpg.0000421407.88128.5c
5. Puertolas MV, Fifi AC. The role of disaccharidase deficiencies in functional abdominal pain disorders-a narrative review. Nutrients. 2018;10(12):1835. https://doi.org/10.3390/nu10121835
6. Daileda T, Baek P, Sutter ME, Thakkar K. Disaccharidase activity in children undergoing esophagogastroduodenoscopy: a systematic review. World J Gastrointest Pharmacol Ther. 2016;7(2):283-293. https://doi.org/10.4292/wjgpt.v7.i2.283
7. Nichols BL Jr, Adams B, Roach CM, Ma CX, Baker SS. Frequency of sucrase deficiency in mucosal biopsies. J Pediatr Gastroenterol Nutr. 2012;55 Suppl 2:S28-S30. https://doi.org/10.1097/01.mpg.0000421405.42386.64
8. Dahlqvist A. Assay of intestinal disaccharidases. Scand J Clin Lab Invest. 1984;44(2):169-172. https://doi.org/10.3109/00365518409161400
9. Cohen SA, Oloyede H, Gold BD, Mohammed A, Elser HE. Clinical characteristics of disaccharidase deficiencies among children undergoing upper endoscopy. J Pediatr Gastroenterol Nutr. 2018;66 Suppl 3:S56-S60. https://doi.org/10.1097/mpg.0000000000001961
10. Haberman Y, Di Segni A, Loberman-Nachum N, et al. Congenital sucrase-isomaltase deficiency: a novel compound heterozygous mutation causing aberrant protein localization. J Pediatr Gastroenterol Nutr. 2017;64(5):770-776. https://doi.org/10.1097/mpg.0000000000001424
11. Uhrich S, Wu Z, Huang JY, Scott CR. Four mutations in the SI gene are responsible for the majority of clinical symptoms of CSID. J Pediatr Gastroenterol Nutr. 2012;55 Suppl 2:S34-S35. https://doi.org/10.1097/01.mpg.0000421408.65257.b5
12. Gericke B, Amiri M, Scott CR, Naim HY. Molecular pathogenicity of novel sucrase-isomaltase mutations found in congenital sucrase-isomaltase deficiency patients. Biochim Biophys Acta Mol Basis Dis. 2017;1863(3):817-826. https://doi.org/10.1016/j.bbadis.2016.12.017
13. Nilholm C, Roth B, Ohlsson B. A dietary intervention with reduction of starch and sucrose leads to reduced gastrointestinal and extra-intestinal symptoms in IBS patients. Nutrients. 2019;11(7):1662. https://doi.org/10.3390/nu11071662
14. Henström M, Diekmann L, Bonfiglio F, et al. Functional variants in the sucrase-isomaltase gene associate with increased risk of irritable bowel syndrome. Gut. 2018;67(2):263-270. https://doi.org/10.1136/gutjnl-2016-312456
15. Deb C, Campion S, Derrick V, et al. Sucrase-isomaltase gene variants in patients with abnormal sucrase activity and functional gastrointestinal disorders. J Pediatr Gastroenterol Nutr. 2021;72(1):29-35. https://doi.org/10.1097/mpg.0000000000002852
16. Dahlqvist A. Disaccharide intolerance. JAMA. 1966;195(3):225-227. doi:10.1001/jama.1966.03100030119039