
Peer Reviewed
Lymphomatoid Granulomatosis Presenting as Cutaneous Lesions in a Patient With Holt-Oram Syndrome
Authors:
Nathan Miller, DO; Tessa Mullins, DO; and Chad Johnston, DO
LewisGale Hospital Montgomery, Blacksburg, Virginia
Citation:
Miller N, Mullins T, Johnston C. Lymphomatoid granulomatosis presenting as cutaneous lesions in a patient with Holt-Oram syndrome. Consultant. 2019;59(5):157-159.
A 33-year-old man with a history of Holt-Oram syndrome (HOS) presented to the clinic with a 10.2 × 7.6-cm necrotic nasal lesion with associated 2.5-cm dome-shaped nodules with central ulceration scattered on his upper extremities, back, torso, and upper thighs (Figures 1 and 2).
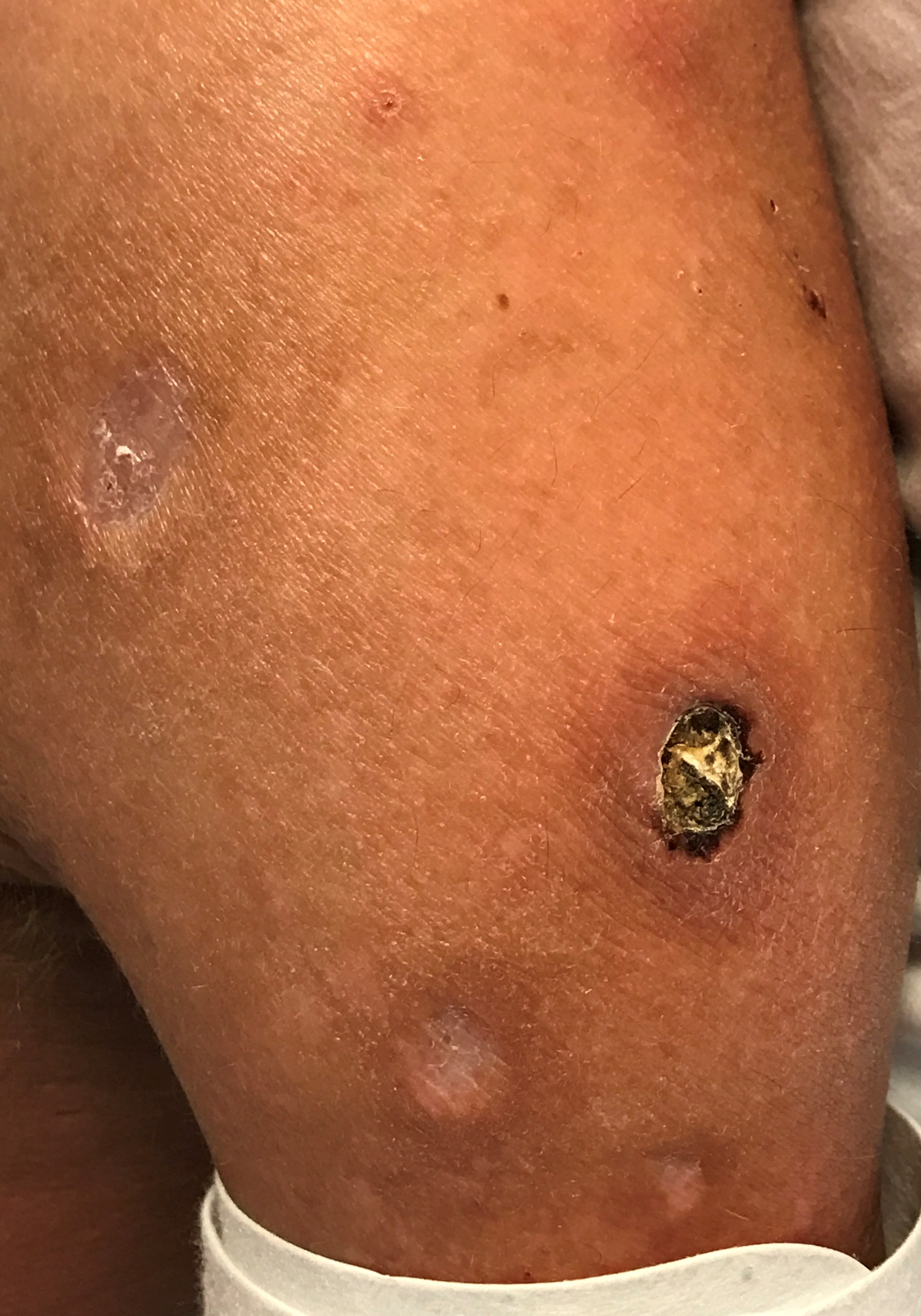
Figure 1. A 10 × 10-cm centrally ulcerated nodule with erythematous borders on the patient’s upper extremity.
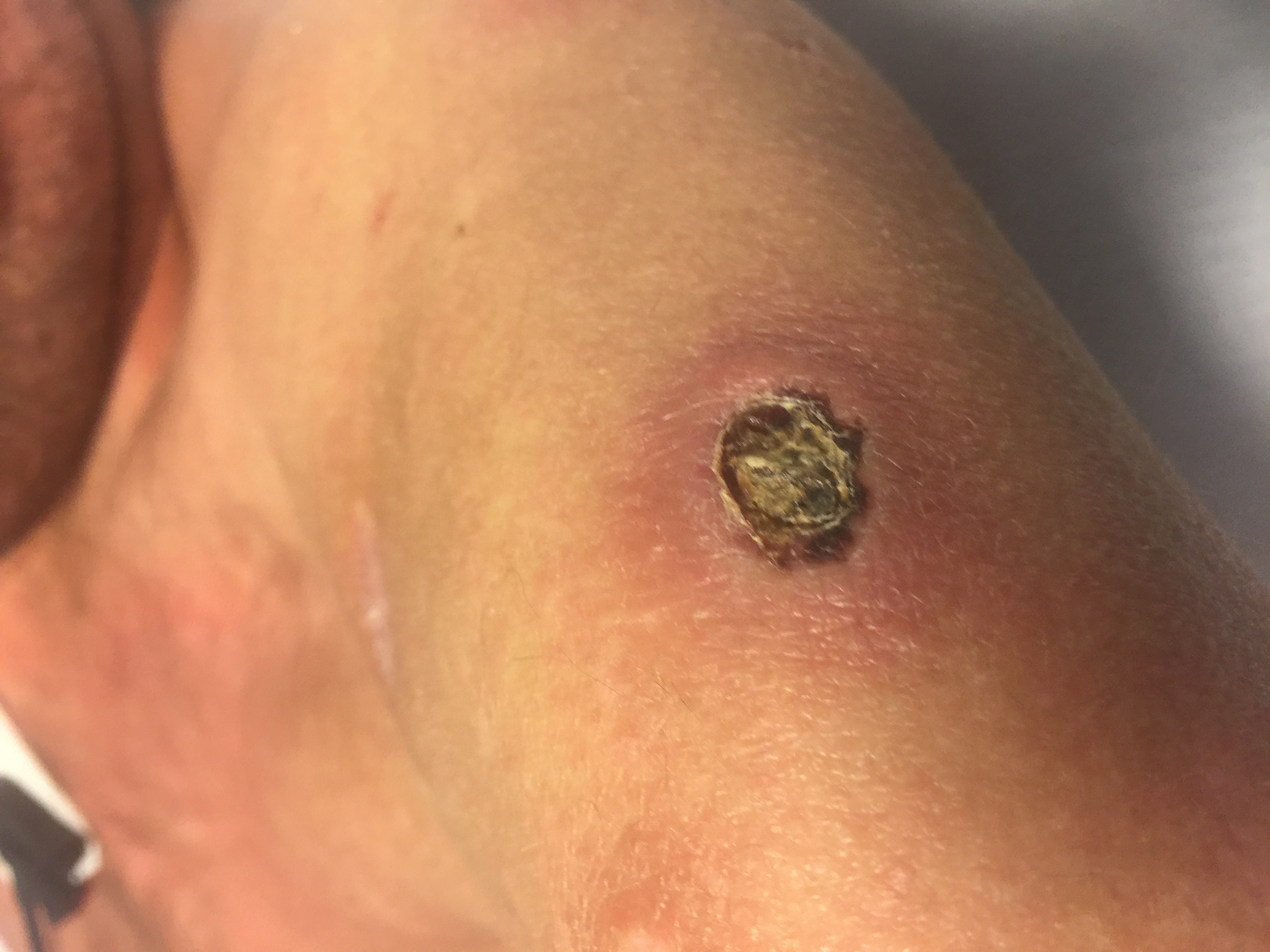
Figure 2. A healing ulcerated lesion on the patient's upper extremity.
He reported that the nasal lesion originally had begun as a small nasal papule 5 months ago, with the scattered nodules appearing 1 month ago. He denied any coughing, dyspnea, or wheezing; there were no abnormal lung findings on physical examination. The patient had had a recent 3-month hospitalization for sepsis secondary to pneumonia.
A biopsy was taken of a lesion on the left arm, with the initial pathology report interpreted as natural killer (NK)/T-cell lymphoma of the midfacial type.
Computed tomography scans of the head and chest showed an axillary nodule with necrosis, a nasopharyngeal mass in the right fossa of Rosenmüller that encased the hard palate, and a right axillary mass with necrosis. His nasal septum was deviated to the left due to the nasopharyngeal mass.
The final pathology report of the skin biopsy showed positive immunohistochemical staining for CD2/3-positive T cells with predominantly infiltrating CD4+ cells. CD20+ cells predominated around blood vessels, with evidence of Epstein-Barr virus (EBV)–positive cells infiltrating the tissue and blood vessels (Figures 3 and 4). Immunohistochemical staining was negative for CD56, ruling out the initial diagnosis of NK/T-cell lymphoma.
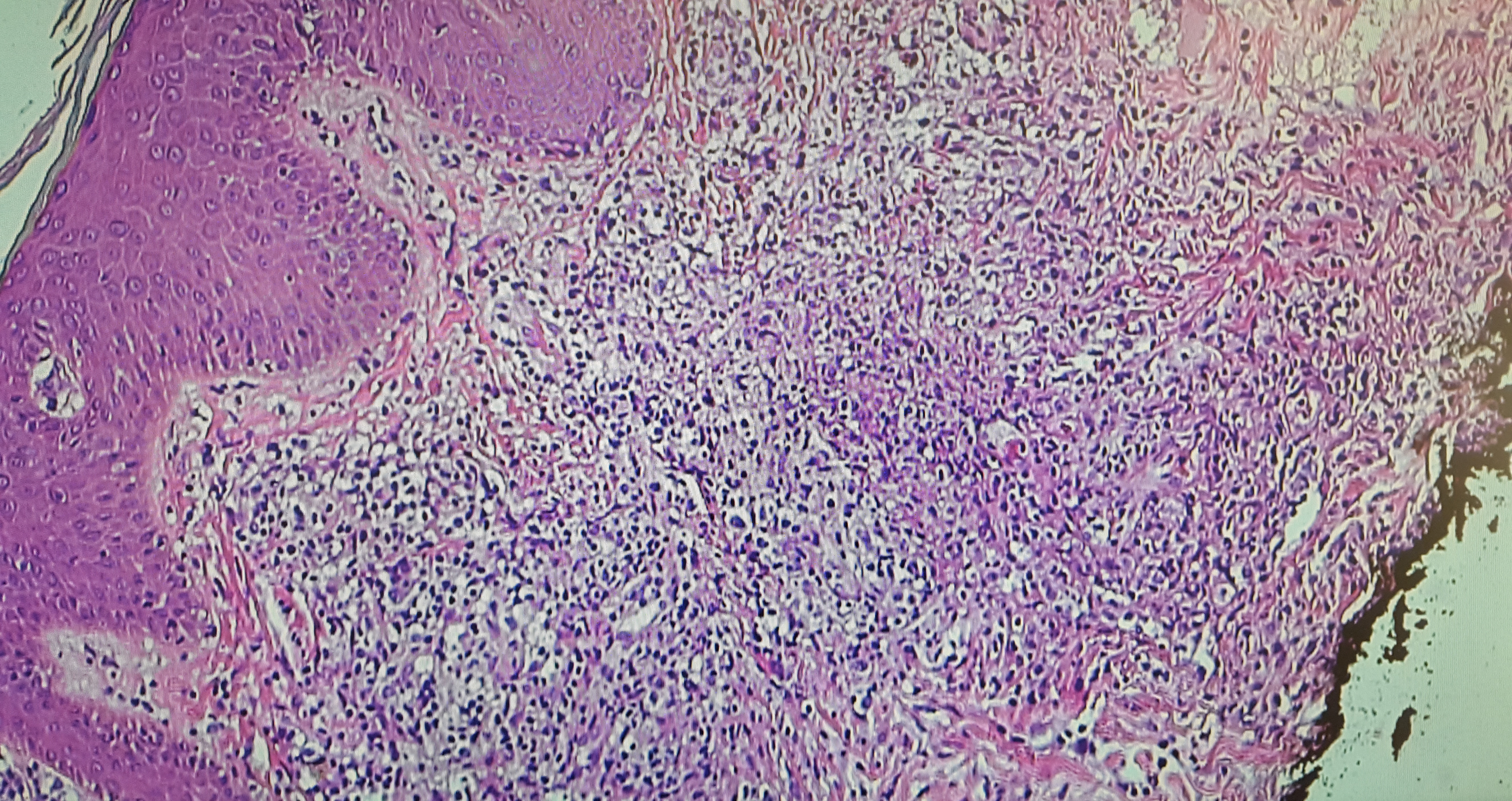
Figure 3. Hematoxylin-eosin stain showing a superficial and deep diffuse inflammatory infiltrate composed of polymorphous lymphocytes.
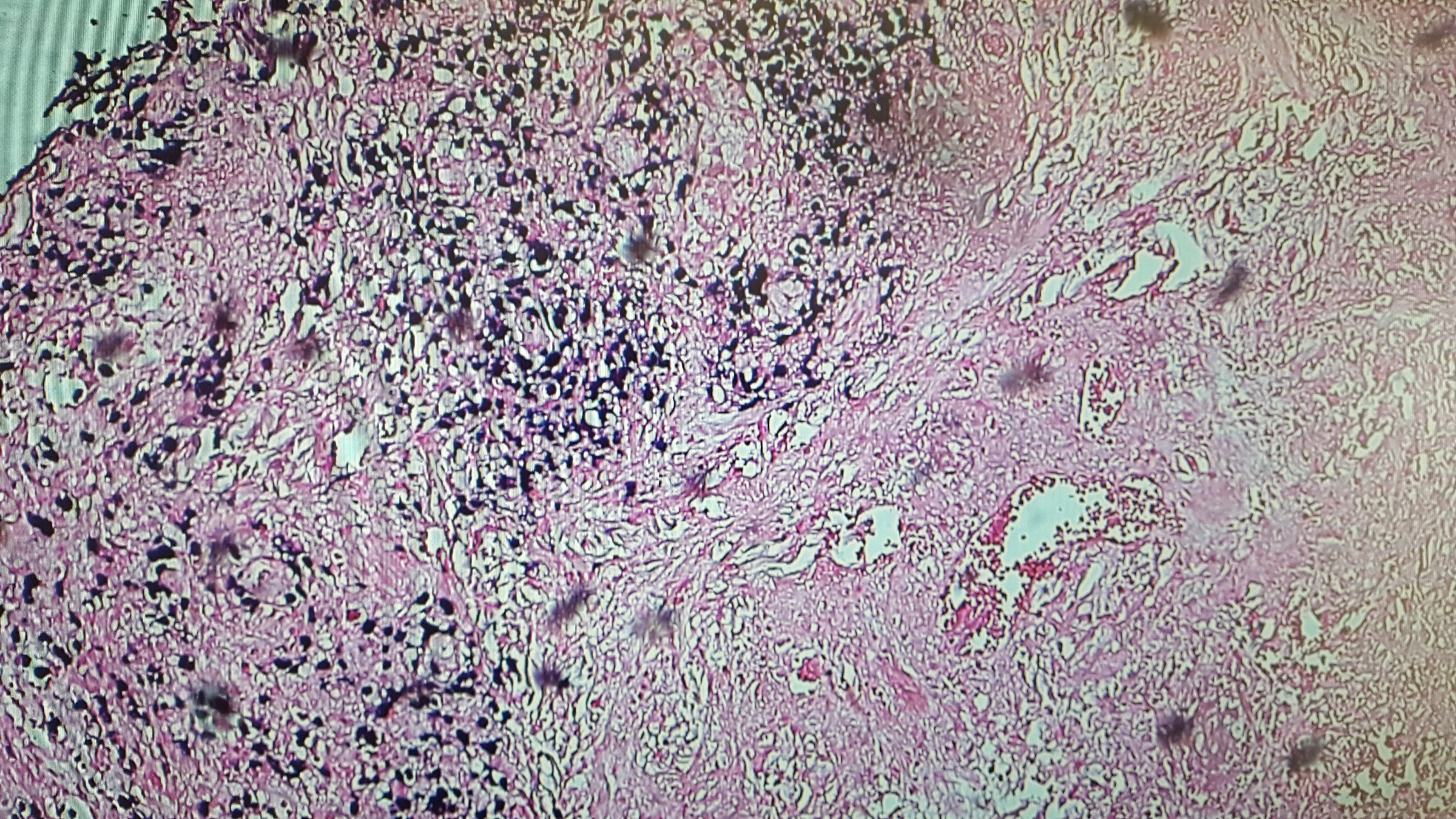
Figure 4. Epstein-Barr encoding region (EBER) in situ hybridization demonstrating positivity for EBV.
The patient received a diagnosis of angiodestructive lymphomatoid granulomatosis (LYG) grade 3. The patient’s laboratory test results showed an EBV immunoglobulin G level of greater than 600 U/mL; results of HIV and hepatitis virus panels were negative.
After further evaluation by an oncologist, the patient also received a diagnosis of EBV-positive diffuse large B-cell lymphoma (DLBCL) and was started on R-EPOCH (rituximab, etoposide phosphate, prednisone, vincristine sulfate, cyclophosphamide, and doxorubicin hydrochloride) chemotherapy, with a goal to complete 6 cycles of treatment. The patient tolerated the first cycles of chemotherapy well and showed clinical improvement.
DISCUSSION
LYG is a rare B-cell disorder that is characterized by overproduction of lymphocytes that infiltrate and accumulate within tissues and around blood vessels, leading to angiodestruction. The signs and symptoms vary greatly and most often include dyspnea, hemoptysis, or cough. Systemic symptoms can also occur and may include fever, malaise, weight loss, and fatigue. LYG most commonly involves the lungs, but cutaneous lesions present in 40% to 50% of patients and vary in presentation. Skin lesions can vary in size and amount of erythema, and they can also ulcerate and become necrotic, as in our patient’s case. Less commonly, the hepatic or renal systems can be involved. Approximately 30% of patients experience central nervous system involvement with sensory or motor defects.
Patients with LYG often have evidence of congenital or acquired immunodeficiency such as HIV disease. It is uncertain whether this patient’s clinical course was complicated by his history of HOS. HOS is an autosomal dominant condition characterized by skeletal abnormalities and, frequently, congenital cardiac defects such as ventricular septal defect and atrial septal defect (ASD).1,2 Our patient reported a history of an ASD that had closed spontaneously during childhood. HOS is not frequently associated with immunodeficiency. In our patient, results of HIV and hepatitis panels were negative, and he denied any sick contacts or acute illness. Of note, he did have a history of multiple respiratory infections in childhood up until adulthood, with his most recent hospitalization for pneumonia only 5 months prior to presentation.
Various treatments are available for LYG. Observation, oral corticosteroids, and chemotherapy have all been used to treat cases of LYG. Although studies have shown no difference in disease-free survival with these treatments, most cases are treated based on clinical presentation and severity. The LYG mortality rate is greater than 60% at 5 years, with the most common cause of death being extensive destruction of pulmonary parenchyma. Patients typically have a median survival rate of 14 months, and the prognosis worsens with higher grade of disease.3
LYG can indolently progress into EBV-positive lymphoma in a minority of cases. EBV is a transforming virus and is able to significantly alter the growth of B cells. EBV binds to the complement receptor CD21 on B cells, resulting in continuous growth.4 Cases of EBV-positive DLBCL have many unfavorable prognostic characteristics regardless of the patient’s age. In one study, EBV-positive lymphomas showed significantly worse overall survival compared with EBV-negative lymphomas.5 EBV-positive DLBCL has a poor response to treatment, so rapid detection is necessary.
This case demonstrates the necessity for immunosuppressed patients to monitor their skin and be aware of acute changes in order to provide an accurate diagnosis and necessary treatment in a timely manner.
- Basson CT, Cowley GS, Solomon SD, et al. The clinical and genetic spectrum of the Holt-Oram syndrome (heart-hand syndrome). N Engl J Med. 1994;330(13):885-89
- Ecob-Newberry RA, Leanage R, Raeburn JA, Young ID. Holt-Oram syndrome: a clinical genetic study. J Med Genet. 1996;33(4):300-307.
- Shaigany S, Weitz NA, Husain S, Geskin L, Grossman ME. A case of lymphomatoid granulomatosis presenting with cutaneous lesions. JAAD Case Rep. 2015;1(4):234-237.
- Nakamura S, Jaffe ES, Swerdlow SH. EBV positive diffuse large B-cell lymphoma of the elderly. In: Swerdlow SH, Campo E, Harris NL, et al, eds. WHO Classification of Tumours of Haematopoietic and Lymphoid Tissues. 4th ed. Lyon, France: International Agency for Research on Cancer; 2008:243-24
- Lu T-X, Liang J-H, Miao Y, et al. Epstein-Barr virus positive diffuse large B-cell lymphoma predict poor outcome, regardless of the age. Sci Rep. 2015;23(5):12168.